
Articles
- Page Path
- HOME > Osong Public Health Res Perspect > Volume 4(6); 2013 > Article
-
Original Article
Lon Mutant ofBrucella abortus Induces Tumor Necrosis Factor-Alpha in Murine J774.A1 Macrophage - Sungdo Park, Young-Sill Choi, Sang-Hee Park, Young-Rok Kim, Hyuk Chu, Kyu-Jam Hwang, Mi-Yeoun Park
-
Osong Public Health and Research Perspectives 2013;4(6):301-307.
DOI: https://doi.org/10.1016/j.phrp.2013.10.002
Published online: October 18, 2013
Division of Zoonoses, Korea National Institute of Health, Osong, Korea
- ∗Corresponding author. miyeoun@korea.kr
• Received: September 9, 2013 • Revised: September 30, 2013 • Accepted: October 2, 2013
© 2013 Published by Elsevier B.V. on behalf of Korea Centers for Disease Control and Prevention.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
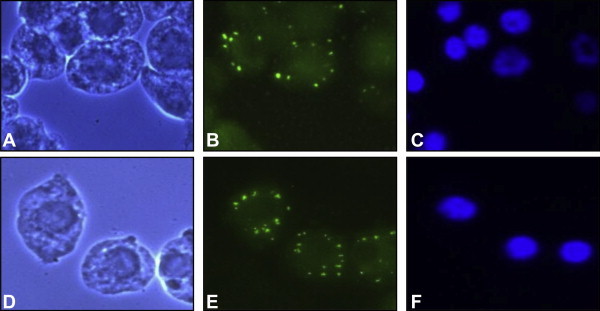
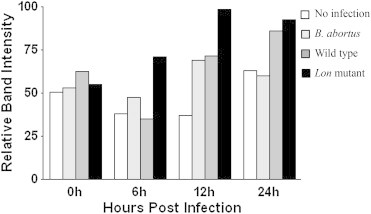
Figure & Data
References
Citations
Citations to this article as recorded by 
- Structure, Substrate Specificity and Role of Lon Protease in Bacterial Pathogenesis and Survival
Perumalraja Kirthika, Khristine Kaith Sison Lloren, Vijayakumar Jawalagatti, John Hwa Lee
International Journal of Molecular Sciences.2023; 24(4): 3422. CrossRef - ClpP protease modulates bacterial growth, stress response, and bacterial virulence in Brucella abortus
Dongjie Sun, Yufu Liu, Xiaowei Peng, Hao Dong, Hui Jiang, Xuezheng Fan, Yu Feng, Jiali Sun, Kun Han, Qiang Gao, Jianrui Niu, Jiabo Ding
Veterinary Research.2023;[Epub] CrossRef - Brucella abortus Encodes an Active Rhomboid Protease: Proteome Response after Rhomboid Gene Deletion
María Inés Marchesini, Ansgar Poetsch, Leticia Soledad Guidolín, Diego J. Comerci
Microorganisms.2022; 10(1): 114. CrossRef - Proteomics of Brucella
Ansgar Poetsch, María Inés Marchesini
Proteomes.2020; 8(2): 8. CrossRef - Outer Membrane Vesicles From Brucella melitensis Modulate Immune Response and Induce Cytoskeleton Rearrangement in Peripheral Blood Mononuclear Cells
Eric Daniel Avila-Calderón, Olín Medina-Chávez, Leopoldo Flores-Romo, José Manuel Hernández-Hernández, Luis Donis-Maturano, Ahidé López-Merino, Beatriz Arellano-Reynoso, Ma. Guadalupe Aguilera-Arreola, Enrico A. Ruiz, Zulema Gomez-Lunar, Sharon Witonsky,
Frontiers in Microbiology.2020;[Epub] CrossRef - RNA-seq reveals the critical role of Lon protease in stress response and Brucella virulence
Yufu Liu, Hao Dong, Xiaowei Peng, Qiang Gao, Hui Jiang, Guanlong Xu, Yuming Qin, Jianrui Niu, Shijing Sun, Peng Li, Jiabo Ding, Ruiai Chen
Microbial Pathogenesis.2019; 130: 112. CrossRef - Brucella Downregulates Tumor Necrosis Factor-α to Promote Intracellular Survival via Omp25 Regulation of Different MicroRNAs in Porcine and Murine Macrophages
Xiaomao Luo, Xiujuan Zhang, Xingchen Wu, Xuefeng Yang, Cong Han, Zhengyu Wang, Qian Du, Xiaomin Zhao, Shan-Lu Liu, Dewen Tong, Yong Huang
Frontiers in Immunology.2018;[Epub] CrossRef
 PubReader
PubReader Cite
Cite


