
Articles
- Page Path
- HOME > Osong Public Health Res Perspect > Volume 10(4); 2019 > Article
-
Original Article
Cell Death Mechanisms in Esophageal Squamous Cell Carcinoma Induced by Vesicular Stomatitis Virus Matrix Protein - Yousef Douzandegana, Alireza Tahamtana,b, Zahra Graya, Hadi Razavi Nikooa, Alijan Tabarraeib, Abdolvahab Moradia
-
Osong Public Health and Research Perspectives 2019;10(4):246-252.
DOI: https://doi.org/10.24171/j.phrp.2019.10.4.08
Published online: August 31, 2019
aDepartment of Microbiology, Faculty of Medicine, Golestan University of Medical Sciences, Gorgan, Iran
bInfectious Diseases Research Centre, Golestan University of Medical Sciences, Gorgan, Iran
- *Corresponding author: Abdolvahab Moradi, Department of Microbiology, Faculty of Medicine, Golestan University of Medical Sciences, Gorgan, Iran, E-mail: abmoradi@gmail.com
• Received: April 24, 2019 • Revised: July 10, 2019 • Accepted: July 28, 2019
Copyright ©2019, Korea Centers for Disease Control and Prevention
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Abstract
-
Objectives
- Vesicular stomatitis virus (VSV) is under development as an oncolytic virus due to its preferential replication in cancer cells and oncolytic activity, however the viral components responsible have not yet been determined. In this study the effects of VSV wild-type (wt) and M51R-mutant matrix proteins (M51R-mMP) on apoptosis, pyroptosis, necroptosis, and autophagy pathways, in an esophagus cancer cell line (KYSE-30) were investigated.
-
Methods
- The KYSE-30 cells were transfected with pcDNA3.1 plasmids encoding wt or M51R-mMP, and apoptosis, pyroptosis, necroptosis, and autophagy were evaluated 48 and 72 hours after transfection.
-
Results
- KYSE-30 cells transfected with VSV wt and M51R-mMPs significantly reduced cell viability to < 50% at 72 hours post-transfection. M51R-MP significantly increased the concentration of caspase-8 and caspase-9 at 48 and 72 hours post-transfection, respectively ( p < 0.05). In contrast, no significant changes were detected following transfection with the VSV wt plasmid. Moreover, VSV wt and M51R-mMP transfected cells did not change the expression of caspase-3. VSV wt and M51R-mMPs did not mMP change caspase-1 expression (a marker of pyroptosis) at 48 and 72 hours post-transfection. However, M51R-mMP and VSV wt transfected cells significantly increased RIP-1 (a marker of necroptosis) expression at 72 hours post-infection ( p < 0.05). Beclin-1, a biomarker of autophagy, was also induced by transfection with VSV wt or M51R-mMPs at 48 hours post-transfection.
-
Conclusion
- The results in this study indicated that VSV exerts oncolytic activity in KYSE-30 tumor cells through different cell death pathways, suggesting that M51R-mMP may potentially be used to enhance oncolysis.
- Oncolytic viruses have gained much attention due to their multimodal mechanism of action [1]. They have an inherent ability to selectively replicate in and lyse tumor cells, and offer a novel approach to stimulate antitumor immune responses [2]. As such, vesicular stomatitis virus (VSV) is being investigated due to its inherent tumor selectivity, rapid replication and cell-killing cycle, as well as its natural ability to stimulate immune responses to break immune tolerance in the tumor microenvironment [3]. Given that, a multitude of strategies have been applied to further improve the safety, selectivity, oncolytic activity, and immune-stimulatory properties of VSV as a novel clinical therapy candidate [4].
- VSV, a member of the Rhabdoviridae family, is a non-segmented negative-strand RNA virus and encodes 5 viral proteins (N, P, M, G and L) [5]. While the virus has the ability to infect a wide range of host cells through targeting receptors, replication is limited to cells that are defective in their antiviral pathways [6]. Tumor cells are defective in antiviral pathways, allowing for an inherent mechanism for tumor specificity for VSV replication [7]. This virus can replicate in immortalized and malignant cells, and eventually induces cell death. Previous studies have also confirmed that VSV can suppress the growth of various tumors upon applying intratumorally or intravenously in tumor-bearing animal models [8].
- Researchers are interested in identifying viral components that play a role in inducing cell death in virus-infected cells [9]. This is important because if the components still maintain this function when used alone, they may be of therapeutic benefit without the potential biological risks of infection with intact virus [10]. Recent studies have introduced the VSV-matrix protein (MP), a structural component of the virion, which causes cytopathogenesis in the absence of other viral components [11]. This includes inhibition of host gene expression at transcription level, mRNA export and translation inhibition, as well as induction of apoptosis [12]. However, there is no data regarding the effects of MP on other cell death pathways.
- There are different cell death pathways including apoptosis, pyroptosis, necroptosis, and autophagy. Apoptosis is programmed cell death that is recognized by a reduction in cell size and morphological changes such as nuclear condensation, fragmentation of DNA and plasma membrane shrinkage. These phenotypic changes result from the action of an effector caspase, caspase-3 or caspase-7, on key cellular molecules [10,13]. Pyroptosis is morphologically and mechanistically distinct from other forms of cell death. Caspase-1 dependence is a defining feature of pyroptosis, and is the enzyme that mediates this process of cell death [14–16]. Necroptosis is a type of regulated cell death dependent on the activity of receptor-interacting serine/threonine-protein (RIP) kinases. However, unlike apoptosis, it is caspase-independent. In addition to their central role in necrosis, the necrosome components receptor-interacting serine/threonine-protein (RIP) 1 and RIP3 have been proposed to be involved in the activation of inflammasomes [17–19]. In mammalian systems, autophagy was recently shown to be important in innate immune defense against intracellular pathogens. Autophagy, or cellular self-digestion, is a cellular pathway crucial for development, differentiation, survival, and homeostasis. One of the main factors regulating autophagy is the human beclin-1 gene, located on chromosome 17q21. Beclin-1 is part of a type III phosphatidylinositol 3-kinase complex required for autophagic vesicle formation [20,21].
- The VSV is a valuable target for oncolytic development and may help with understanding the role of viral components in the induction of the cell death pathways for the development of effective therapies. This study investigated the potential anti-tumor effects of VSV wild type (wt) and M51R-mutant matrix protein (mMP) on apoptosis, pyroptosis, necroptosis and autophagy in esophageal squamous cell carcinoma (SCC) (S-30).
Introduction
- 1. Cell line, plasmids and transfection
- The in vitro study was performed in the esophagus cancer cell line, KYSE-30. The cells were cultured in Roswell park memorial institute medium (RPMI)-1640 medium (Gibco, UK) supplemented with 10% fetal bovine serum (Gibco, UK), 100 u/mL penicillin and 100 μg/mL streptomycin at pH 7.2 at 37°C with 5% CO2.
- The pcDNA3.1 plasmids expressing VSV (wild-type) wt and M51R-mMP were generated. For construction of the pcDNA3.1 plasmid expressing VSV wt MP, the cDNA sequences of the M gene were cloned into the plasmid. To generate pcDNA3.1-M51R-mMP, the methionine was changed at position 51 to arginine. Bacterial strain Escherichia coli DH5α (Pasteur Institute of Iran) was used for propagation and preparation of constructed plasmids.
- KYSE-30 cells with a density of 106 cells/well were seeded onto 6-well plates for 24 hours in 3 mL of growth medium. Before transfection, the medium was removed and the cells were washed with PBS and then replaced with fresh culture medium without fetal bovine serum. The cells were incubated with 250 μL of the plasmids (5 μg) and lipofectamine 2000 reagent (Invitrogen) (6 μL) and maintained for 6 hours at 37°C under 5% CO2. The plasmids and lipofectamine 2,000 solutions were diluted with serum and antibiotic-free RPMI-1640 separately to reach a final volume of 250 μL. Finally, the transfection solution was aspirated and replaced with 1 mL of complete medium per well. In the present study, the optimum amount of DNA for transfection was obtained from a previous study [22].
- 2. Experimental assays
- The expression of VSV wt and M51R-mMPs from encoding plasmids, 48 and 72 hours post-transfection, was performed by SDS-PAGE and Western blotting. In SDS-PAGE, the extracted lysate total proteins in the sample loading buffer, were separated by PAGE, and stained by coomassie blue R250. For blotting, proteins separated by SDS-PAGE were transferred onto nitrocellulose membrane (Roche, Germany), and hybridized with the monoclonal anti-VSV MP antibody 23H12 (1:5,000). Detection was performed with enhanced chemiluminescence reagents according to instructions.
- In order to evaluate cell viability, KYSE-30 cells were cultivated on 96-well plates at a density of 5×104 cells/well in 150 μL fresh culture medium supplemented with 10% fetal bovine serum. Cell viability was performed with an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] kit according to manufacturer’s instruction (Roche, Germany).
- To analyze the death pathway induction at 48 and 72 hours after transfection, cell lysates were examined by Enzyme Linked Immunosorbent Assay kits according to manufacturer’s instruction. The following markers were analyzed: caspase-9 (Abnova, Taiwan), caspase-8 (Abcam, UK), caspase-3 (Abcam, UK), caspase-1 (Abnova, Taiwan), beclin-1 (LSBio, USA), and RIP1 (LSBio, USA).
- 3. Statistical analysis
- Data analyses were performed via GraphPad prism (GraphPad Software, Inc., La Jolla, CA, USA). Differences were analyzed through ANOVA test, followed by Bonferonni’s post hoc test. A value of p less than 0.05 was considered to be statistically significant.
Materials and Methods
- To evaluate the expression of VSV wt and M51R-mMPs in transfected KYSE-30 cells, Western blot analysis was performed. The housekeeping gene beta-actin was used as control. As shown in Figure 1A, the constructed plasmids expressed the VSV wt and M51R-mMP proteins.
- After transfection of VSV wt and M51R-mMPs plasmids into the KYSE-30 cells, the MTT assay was used to determine cell viability at 24, 48 and 72 hours after induction. The results showed that KYSE-30 cells transfected with VSV wt and M51R-mMPs significantly reduced cell viability to < 50% at 72 hours post-transfection (Figure 1B).
- The apoptosis pathway was assessed by analysis for the expression of caspase-9, caspase-8 and caspase-3 at 48 and 72 hours post-transfection. The data showed that VSV M51R-MP statistically significantly increased the concentration of caspase-8 and caspase-9 at 48 and 72 hours post-transfection, respectively (p < 0.05). No statistically significant changes were detected following transfection with the VSV wt plasmid (Figures 2A and 2B). Moreover, VSV wt and M51R-mMP transfected cells did not change the expression of caspase-3 (Figure 2C).
- Pyroptosis was investigated by measuring the level of caspase-1 expression. As shown in Figure 3, VSV wt and M51R-mMPs did not change caspase-1 expression at 48 and 72 hours post-transfection. RIP-1 expression level was measured to determine the necroptosis induction. M51R-mMP significantly increased RIP-1 expression at 48 and 72 hours post-infection (p < 0.05). Furthermore, the VSV wt transfected cells significantly increased the expression of RIP-1 at 72 hours post-infection (p < 0.05; Figure 4). Beclin-1 expression measured as a biomarker for autophagy induction, was significantly increased at 48 hours post-transfection by VSV wt and M51R-mMPs compared with the plasmid vector only control (Figure 5).
Results
- Esophageal cancer remains a major public health problem worldwide. Chemotherapy, radiotherapy, novel targeted molecular drugs therapy, and immune checkpoint inhibitors, together present new therapeutic approaches for patients with SCCs in the near future [23]. However, oncolytic viruses are potential physiological agents for cancer cell treatment.
- Understanding the different mechanisms of cell death is crucial for cancer therapy success. VSV is an oncolytic virus that selectively infects and kills cancer cells but how VSV induces cell death is not fully determined. This study investigated the effect of VSV wt and M51R-mMPs on KYSE-30 cell death pathways. Identification of the viral components involved in inducing cell death would enable testing of these components alone, compared with the production of these components from the virus. Potentially any proteins causing cell death could be expressed using vectors as therapies, thereby, avoiding any potential biological risks of using intact VSV.
- In this study, it was determined that both wt and M51R-mMPs were capable of inducing cell death pathways, especially via autophagy. However, in this study there was a reduction in the expression of cell death markers by wt MPs compared with M51R-mMPs. It has been reported that wt MPs-induced inhibition of host nuclear-cytoplasmic RNA transport, which has been attributed to the interaction of wt MP with a nuclear pore component (nucleoporin Nup98) [24,25].
- The recognition of VSV G glycoprotein by Toll-like receptor 4, and signaling through this receptor can activate autophagy directly [26–28] and the recognition of VSV infection is thought to involve toll-like receptor (TLR) 13 [29]. Furthermore, nuclear factor erythroid 2-related factor 2, has a role in the regulation of autophagy which complements the therapeutic potential of VSV oncolysis [30,31]. Although, it has been suggested that VSV-mediated autophagy occurs via interaction of VSV G glycoprotein with TLR-4 [32,33], the role of VSV-MP has not yet been defined. This is the first report of Beclin1-mediated autophagy activation with both VSV wt and M51R-mMPs in SCCs. Post-transcriptional modifications on the autophagy proteins such as Beclin1, autophagy-related proteins, resulted in the formation of autophagosomes and fine-tuning of the responses to autophagy stimulus [34]. Hence, Beclin1 phosphorylation is supposed to be a central mediator of autophagy regulation and also mediate the cross-talk between autophagy and necroptosis responses [35].
- In several models, autophagy has been shown to regulate necroptosis [36,37]. Treatment with N-benzyloxycarbonyl-Valyl-Alanyl-Aspartyl-fluoromethylketone, a caspase inhibitor with broad specificity, induced autophagy and the death of mouse subcutaneous connective tissue via engagement of RIP1 [38]. In the present study, RIP1 was increased by both VSV wt and M51R-mMP. Wang et al, showed that VSV infection triggered the RIP1-RIP3 complex which is an activator to form necroptosis [39], supporting the results from this current study. RIP1 is an important regulatory protein in the death-inducing signaling complex that can activate NF-κB and caspase-8 and generate reactive oxygen species [40]. A gene knockout of RIP1 suppresses the expression of microtubule-associated protein 1 light chain 3-II, which is the hallmark of autophagy. In addition, loss of RIP1 suppresses Beclin1, which is a substrate of caspase-8, during death receptor-mediated autophagy in macrophages [41]. The results in this current study showed caspase-8 induction by both wt and M51R-mMPs. Studies have shown that a slight increase in caspase-8 not only prevents induction of apoptosis, but also contributes to the progression of the necrotic pathway [42]. Thus, low caspase-8 may benefit from therapies designed to bypass apoptosis and induce necroptosis [43].
- The results from this current study showed that both wt and M51R-mMPs did not activate caspase-1, a non-apoptotic caspase. Previous studies suggest that macrophages activate autophagy in parallel with inflammasome activation, as a means to delay the onset of pyroptosis [44]. Moreover, depletion of autophagic proteins, LC3B (microtubule associated protein-1 light chain 3B) and Beclin1, enhances caspase-1 activation [45]. Although Poeck et al, proposed that VSV is detected by a novel retinoic acid inducible gene 1 protein/apoptosis-associated speck-like protein/caspase-1 inflammasome that does not require nucleotide-binding domain, leucine-rich-containing family (NLRP3) [46]. Nevertheless, further work indicated a requirement for NLRP3 in response to both encephalomyocarditis virus and VSV [47]. Our study suggests that autophagic proteins induced by VSV matrix proteins (MP), regulated NALP3-dependent inflammation. Further work will be required to better delineate how RNA viruses impact on the inflammasome.
- The data in this current study also showed activation of caspase-9 and caspase-8 as apoptotic biomarkers for intrinsic and extrinsic pathways, respectively, by VSV wt and M51R-mMPs, but surprisingly caspase-3, which is involved in apoptosis pathways, was not activated in KYSE-30 cells. These results indicate that apoptosis inhibitors may have a role in this observation. Although VSV MP-induced proteasomal elimination of myeloid cell leukemia 1 protein (Mcl-1) contributes to VSV-induced apoptosis, but not to VSV-induced autophagy [48]. However, the interaction between Beclin1 with Bcl-xL and B-cell lymphoma 2 (Bcl-2) proteins, repressed the intrinsic pathway of apoptosis [49–51]. In addition, Bcl-2 inhibits Fas-induced apoptosis by preventing the formation of the death-inducing complex which is composed of Fas, Fas-associated protein with death domain and Caspase 8 [52].
- In conclusion, we showed the physiological relevance of autophagic regulation in response to VSV MPs in SCC cells. Our data confirmed that apoptosis is not only a cell death pathway that was induced by VSV MPs, but also different cell types have different strategies for cell death pathway activation. However, it is unclear how this observation represented increased autophagic activity in KYSE-30 through VSV MPs. These results suggest a novel role for VSV MPs, as a promising new therapeutic strategy. Particularly, VSV wt or M51R-mMPs could be used in viral vectors for effective oncolytic treatment. This study was limited as more detailed evaluation of mRNA transcription of cell death pathway biomarkers could have been performed. In addition, comparing the effects of VSV MP and M51R-mMPs in different cell lines would have provided more comprehensive results. Moreover, there was no data regarding other cell death pathways taken into account that limitated the current study.
Discussion
-
Acknowledgements
- This work was supported by Golestan University of Medical Sciences (No.: 940819199).
Acknowledgments
- 1. Lin CZ, Xiang GL, Zhu XH, et al. Advances in the mechanisms of action of cancer-targeting oncolytic viruses. Oncol Lett 2018;15(4). 4053−60. PMID: 29541169. PMID: 5835897.ArticlePubMedPMC
- 2. Melzer M, Lopez-Martinez A, Altomonte J. Oncolytic vesicular stomatitis virus as a viro-immunotherapy: Defeating cancer with a “Hammer” and “Anvil”. Biomedicines 2017;5(1). 8PMID: 10.3390/biomedicines5010008. PMID: 5423493.ArticlePubMedPMC
- 3. Bishnoi S, Tiwari R, Gupta S, et al. Oncotargeting by Vesicular Stomatitis Virus (VSV): Advances in Cancer Therapy. Viruses 2018;10(2). 90PMID: 10.3390/v10020090. PMID: 5850397.ArticlePubMedPMC
- 4. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci 2016;107(10). 1373−9. PMID: 10.1111/cas.13027. PMID: 27486853. PMID: 5084676.ArticlePubMedPMC
- 5. Dietzgen RG, Kondo H, Goodin MM, et al. The family Rhabdoviridae: mono-and bipartite negative-sense RNA viruses with diverse genome organization and common evolutionary origins. Virus Res 2017;227:158−70. PMID: 10.1016/j.virusres.2016.10.010.ArticlePubMed
- 6. Hastie E, Cataldi M, Marriott I, et al. Understanding and altering cell tropism of vesicular stomatitis virus. Virus Res 2013;176(1–2). 16−32. PMID: 10.1016/j.virusres.2013.06.003. PMID: 23796410. PMID: 3865924.ArticlePubMed
- 7. Noser JA, Mael AA, Sakuma R, et al. The RAS/Raf1/MEK/ERK signaling pathway facilitates VSV-mediated oncolysis: implication for the defective interferon response in cancer cells. Mol Ther 2007;15(8). 1531−6. PMID: 10.1038/sj.mt.6300193. PMID: 17505473.ArticlePubMed
- 8. Felt SA, Grdzelishvili VZ. Recent advances in vesicular stomatitis virus-based oncolytic virotherapy: a 5-year update. J Gen Virol 2017;98(12). 2895−911. PMID: 10.1099/jgv.0.000980.ArticlePubMedPMC
- 9. _Kaminskyy V, Zhivotovsky B. To kill or be killed: how viruses interact with the cell death machinery. J Intern Med 2010;267(5). 473−82. PMID: 10.1111/j.1365-2796.2010.02222.x. PMID: 20433575.ArticlePubMed
- 10. Danthi P. Viruses and the diversity of cell death. Annu Rev Virol 2016;3(1). 533−53. PMID: 10.1146/annurev-virology-110615-042435. PMID: 27501259.ArticlePubMed
- 11. Publicover J, Ramsburg E, Robek M, et al. Rapid pathogenesis induced by a vesicular stomatitis virus matrix protein mutant: viral pathogenesis is linked to induction of tumor necrosis factor alpha. J Virol 2006;80(14). 7028−36. PMID: 10.1128/JVI.00478-06. PMID: 16809308. PMID: 1489072.ArticlePubMedPMC
- 12. Redondo N, Madan V, Alvarez E, et al. Impact of vesicular stomatitis virus M proteins on different cellular functions. PloS One 2015;10(6). e0131137PMID: 10.1371/journal.pone.0131137. PMID: 26091335. PMID: 4474437.ArticlePubMedPMC
- 13. Ghavami S, Hashemi M, Ande SR, et al. Apoptosis and cancer: mutations within caspase genes. J Med Genet 2009;46(8). 497−510. PMID: 10.1136/jmg.2009.066944. PMID: 19505876.ArticlePubMed
- 14. de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, et al. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ 2019;26(1). 146−61. PMID: 10.1038/s41418-018-0106-7.ArticlePubMedPDF
- 15. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 2009;7(2). 99−109. PMID: 10.1038/nrmicro2070. PMID: 19148178. PMID: 2910423.ArticlePubMedPMCPDF
- 16. Nagarajan K, Soundarapandian K, Thorne RF, et al. Activation of Pyroptotic Cell Death Pathways in Cancer: An Alternative Therapeutic Approach. Transl Oncol 2019;12(7). 925−31. PMID: 10.1016/j.tranon.2019.04.010. PMID: 31085408. PMID: 6518321.ArticlePubMedPMC
- 17. Degterev A, Ofengeim D, Yuan J. Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad Sci USA 2019;116(20). 9714−22. PMID: 31048504. PMID: 6525537.ArticlePubMedPMC
- 18. Vandenabeele P, Galluzzi L, Vanden Berghe T, et al. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010;11(10). 700−14. PMID: 10.1038/nrm2970. PMID: 20823910.ArticlePubMedPDF
- 19. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature 2015;517(7534). 311−20. PMID: 10.1038/nature14191. PMID: 25592536.ArticlePubMedPDF
- 20. Condello M, Pellegrini E, Caraglia M, et al. Targeting Autophagy to Overcome Human Diseases. Int J Mol Sci 2019;20(3). 725PMID: 10.3390/ijms20030725. PMID: 6387456.ArticlePubMedPMC
- 21. Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol 2013;15(7). 713−20. PMID: 10.1038/ncb2788. PMID: 23817233.ArticlePubMedPMCPDF
- 22. Douzandegan Y, Gray Z, Mohebbi A, et al. Optimization of KYSE-30 Esophagus Cancer Cell Line Transfection Using Lipofectamine 2000. J Clin Basic Res 2017;1(2). 16−20.
- 23. Tanaka Y, Yoshida K, Suetsugu T, et al. Recent advancements in esophageal cancer treatment in Japan. Ann Gastroenterol 2018;2(4). 253−65. PMID: 10.1002/ags3.12174.Article
- 24. Ahmed M, McKenzie MO, Puckett S, et al. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J Virol 2003;77(8). 4646−57. PMID: 10.1128/JVI.77.8.4646-4657.2003. PMID: 12663771. PMID: 152115.ArticlePubMedPMC
- 25. Von Kobbe C, van Deursen JM, Rodrigues JP, et al. Vesicular stomatitis virus matrix protein inhibits host cell gene expression by targeting the nucleoporin Nup98. Mol Cell 2000;6(5). 1243−52. PMID: 10.1016/S1097-2765(00)00120-9. PMID: 11106761.ArticlePubMed
- 26. Georgel P, Jiang Z, Kunz S, et al. Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology 2007;362(2). 304−13. PMID: 10.1016/j.virol.2006.12.032. PMID: 17292937.ArticlePubMed
- 27. Xu Y, Jagannath C, Liu X-D, et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007;27(1). 135−44. PMID: 10.1016/j.immuni.2007.05.022. PMID: 17658277. PMID: 2680670.ArticlePubMedPMC
- 28. Lee HK, Lund JM, Ramanathan B, et al. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007;315(5817). 1398−401. PMID: 10.1126/science.1136880. PMID: 17272685.ArticlePubMed
- 29. Shi Z, Cai Z, Sanchez A, et al. A novel Toll-like receptor that recognizes vesicular stomatitis virus. J Biol Chem 2011;286(6). 4517−24. PMID: 10.1074/jbc.M110.159590. PMID: 3039399.ArticlePubMed
- 30. _Ramezani A, Nahad MP, Faghihloo E. The role of Nrf2 transcription factor in viral infection. J Cell Biochem 2018;119(8). 6366−82. PMID: 10.1002/jcb.26897. PMID: 29737559.ArticlePubMed
- 31. Olagnier D, Lababidi RR, Hadj SB, et al. Activation of Nrf2 signaling augments vesicular stomatitis virus oncolysis via autophagy-driven suppression of antiviral immunity. Mol Ther 2017;25(8). 1900−16. PMID: 10.1016/j.ymthe.2017.04.022. PMID: 28527723. PMID: 5542709.ArticlePubMedPMC
- 32. Nabizadeh A, Bamdad T, Arefian E, et al. Autophagy gene activity may act as a key factor for sensitivity of tumor cells to oncolytic vesicular stomatitis virus. Iran J Cancer Prev 2016;9(1). e3919PMID: 10.17795/ijcp-3919. PMID: 27366311. PMID: 4922204.ArticlePubMedPMC
- 33. Shelly S, Lukinova N, Bambina S, et al. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 2009;30(4). 588−98. PMID: 10.1016/j.immuni.2009.02.009. PMID: 19362021. PMID: 2754303.ArticlePubMedPMC
- 34. Menon MB, Dhamija S. Beclin 1 phosphorylation–at the centre of autophagy regulation. Front Cell Dev Biol 2018;6:137PMID: 10.3389/fcell.2018.00137.ArticlePubMedPMC
- 35. Ryter SW, Mizumura K, Choi AM. The impact of autophagy on cell death modalities. Int J Cell Biol 2014;2014:502676. PMID: 10.1155/2014/502676. PMID: 24639873. PMID: 3932252.ArticlePDF
- 36. Bell BD, Leverrier S, Weist BM, et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci USA 2008;105(43). 16677−82. PMID: 10.1073/pnas.0808597105. PMID: 18946037. PMID: 2575479.ArticlePubMedPMC
- 37. Khan MJ, Alam MR, Waldeck-Weiermair M, et al. Inhibition of autophagy rescues palmitic acid-induced necroptosis of endothelial cells. J Biol Chem 2012;287(25). 21110−20. PMID: 10.1074/jbc.M111.319129. PMID: 22556413. PMID: 3375534.ArticlePubMedPMC
- 38. Yu L, Alva A, Su H, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004;304(5676). 1500−2. PMID: 10.1126/science.1096645. PMID: 15131264.ArticlePubMed
- 39. Wang X, Jiang W, Yan Y, et al. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat Immunol 2014;15(12). 1126−33. PMID: 10.1038/ni.3015. PMID: 25326752.ArticlePubMedPDF
- 40. Kelliher MA, Grimm S, Ishida Y, et al. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 1998;8(3). 297−303. PMID: 10.1016/S1074-7613(00)80535-X. PMID: 9529147.ArticlePubMed
- 41. Yao Z, Zhang P, Guo H, et al. RIP1 modulates death receptor mediated apoptosis and autophagy in macrophages. Mol Oncol 2015;9(4). 806−17. PMID: 10.1016/j.molonc.2014.12.004. PMID: 25583602. PMID: 5528779.ArticlePubMed
- 42. _Tummers B, Green DR. Caspase-8: regulating life and death. Immunol Rev 2017;277(1). 76−89. PMID: 10.1111/imr.12541. PMID: 28462525. PMID: 5417704.ArticlePubMedPMC
- 43. Feltham R, Vince JE, Lawlor KE. Caspase-8: not so silently deadly. Clin Transl Immunology 2017;6(1). e124PMID: 10.1038/cti.2016.83. PMID: 28197335. PMID: 5292560.ArticlePubMedPMC
- 44. Byrne BG, Dubuisson J-F, Joshi AD, et al. Inflammasome components coordinate autophagy and pyroptosis as macrophage responses to infection. MBio 2013;4(1). e00620−12. PMID: 10.1128/mBio.00620-12. PMID: 23404401. PMID: 3573666.ArticlePubMedPMC
- 45. Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011;12(3). 222−30. PMID: 10.1038/ni.1980. PMID: 3079381.ArticlePubMedPDF
- 46. Poeck H, Bscheider M, Gross O, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1β production. Nat Immunol 2010;11(1). 63−9. PMID: 10.1038/ni.1824.ArticlePubMedPDF
- 47. Rajan JV, Rodriguez D, Miao EA, et al. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J Virol 2011;85(9). 4167−72. PMID: 10.1128/JVI.01687-10. PMID: 21289120. PMID: 3126243.ArticlePubMedPMC
- 48. Schache P, Gürlevik E, Strüver N, et al. VSV virotherapy improves chemotherapy by triggering apoptosis due to proteasomal degradation of Mcl-1. Gene Ther 2009;16(7). 849−61. PMID: 10.1038/gt.2009.39. PMID: 19369968.ArticlePubMedPDF
- 49. Fernández ÁF, Sebti S, Wei Y, et al. Disruption of the beclin 1–BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018;558(7708). 136−40. PMID: 10.1038/s41586-018-0162-7. PMID: 29849149. PMID: 5992097.ArticlePubMedPMCPDF
- 50. Marquez RT, Xu L. Bcl-2: Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res 2012;2(2). 214−21. PMID: 22485198. PMID: 3304572.PubMedPMC
- 51. Lindqvist LM, Heinlein M, Huang DC, et al. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc Natl Acad Sci USA 2014;111(23). 8512−7. PMID: 10.1073/pnas.1406425111. PMID: 24912196. PMID: 4060681.ArticlePubMedPMC
- 52. _Kawahara A, Kobayashi T, Nagata S. Inhibition of Fas-induced apoptosis by Bcl-2. Oncogene 1998;17(20). 2549−54. PMID: 10.1038/sj.onc.1202192. PMID: 9840917.ArticlePubMedPDF
References
Figure 1
VSV wt and M51R-MP expression in KYSE-30 cell line.
(A) Protein lysates from uninfected and transfected (pcDNA3.1 VSV wt-MP and pcDNA3.1+VSV M51R-mMP) KYSE-30 cells were separated by SDS-PAGE along with total protein at 48 and 72 hours -and transferred to nitrocellulose membrane. VSV wt-MP and M51R-MP were detected as 26 kilodaltons with ECL reagents kit by autoradiography. (B) MTT Assay showing the effect of wild-type Matrix Protein and M51R-Matrix Protein on KYSE-30 cell line. After transfection, cell viability was examined at 24, 48 and 72 hours. Data showed that both MP and M51R-MP significantly reduced cell viability to < 50% at 72 hours post-transfection, compared to untreated cells. ** p < 0.001.
VSV = vesicular stomatitis virus; MP = matrix protein; wt = wild-type; M51R-mMP = M51R-mutant matrix protein; ECL = enhanced chemiluminescence; MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
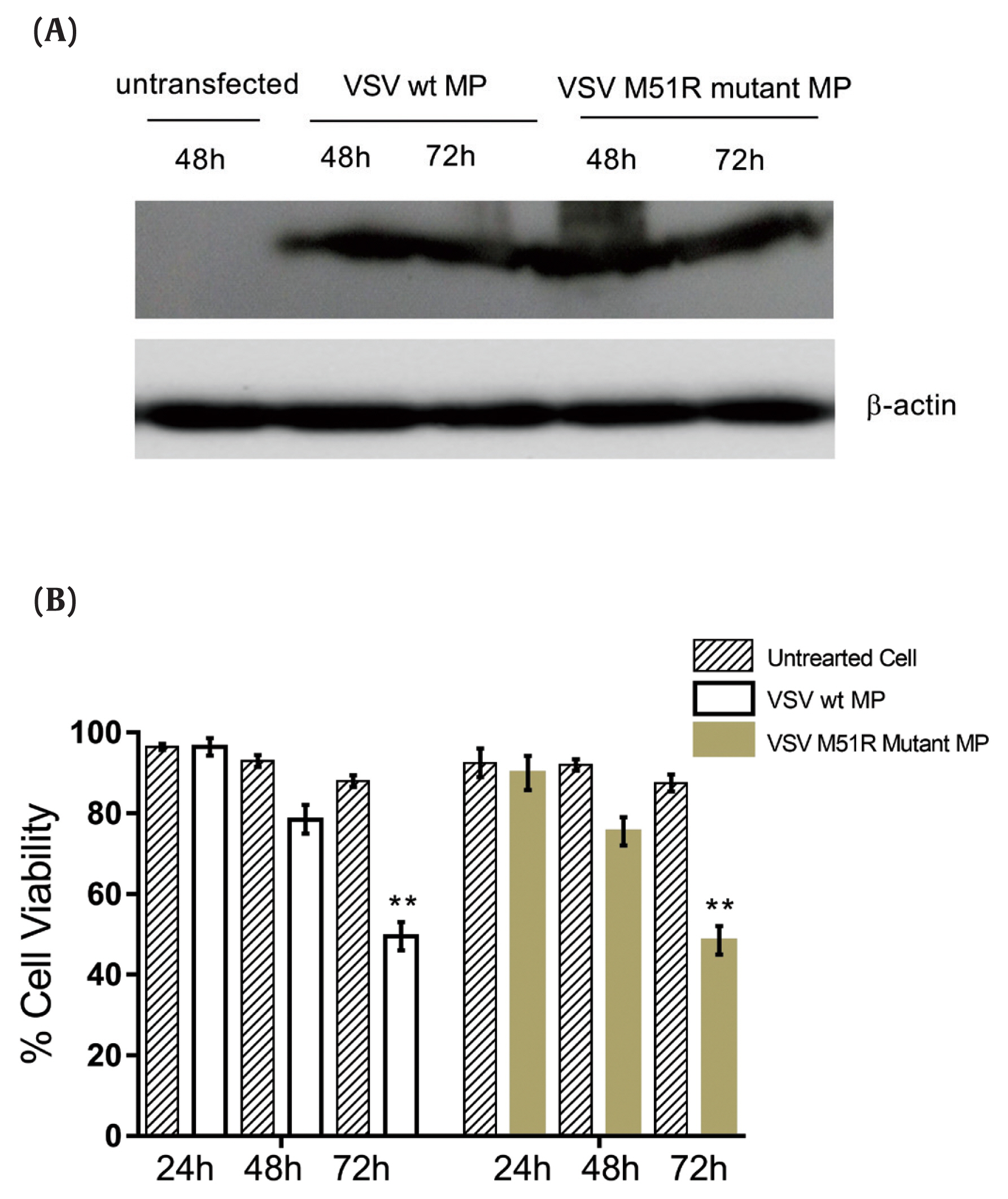
Figure 2
Transfection with wt and M51R-mMP induced intrinsic and extrinsic pro-apoptotic pathways in KYSE-30 cells.
(A) Caspase-8, (B) caspase-9 and (C) caspase-3 were assayed in KYSE-30 cells that untransfected or transfected with pcDNA3.1, wt and M51R-mMP for 48 (black bars) and 72 (Mustard bars) h post-transfection. Expressing wt and M51R-mMP has not potent to induced effector apoptotic protein in KYSE-30 cells. **p < 0.001, ***p < 0.0001.
wt = wild-type; M51R-mMP = M51R-mutant matrix protein.
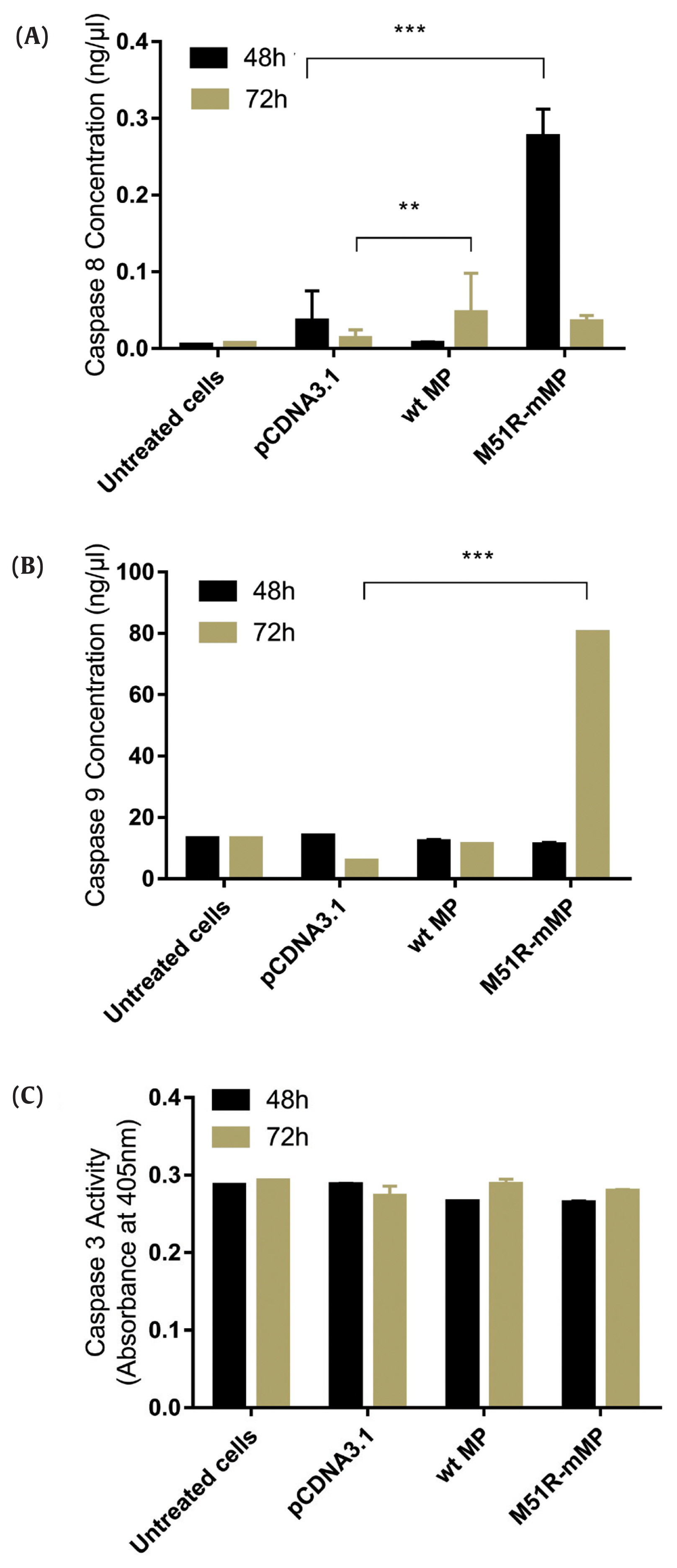
Figure 3
Expression level of Caspase-1. Caspase-1 was assayed in KYSE-30 cells that were untransfected or transfected with pcDNA3.1, wt and M51R-mMP for 48 (black bars) and 72 (Mustard bars) h post-transfection.
wt = wild-type; M51R-mMP = M51R-mutant matrix protein.
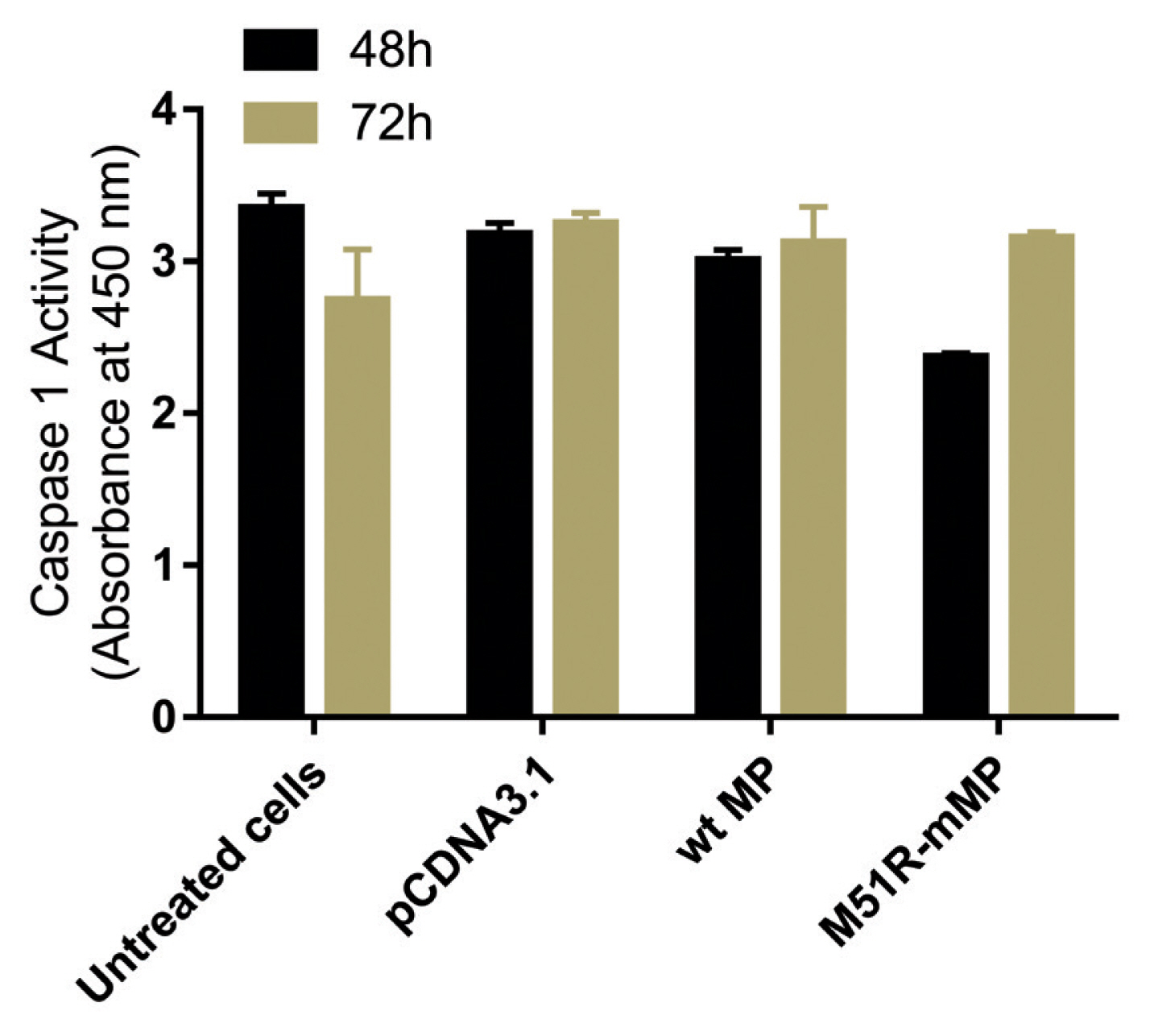
Figure 4
Expression of necroptotic proteins in KYSE-30 cells transfected with wt and M51R-mMP.
RIP1 was assayed in KYSE-30 cells that were untransfected or transfected with pcDNA3.1, wt. or M51R-mMP at 48 (black bars) and 72 (mustard bars) hours post-transfection. *** p < 0.0001.
wt = wild-type; M51R-mMP = M51R-mutant matrix protein; RIP1 = receptor-interacting serine/threonine-protein 1.
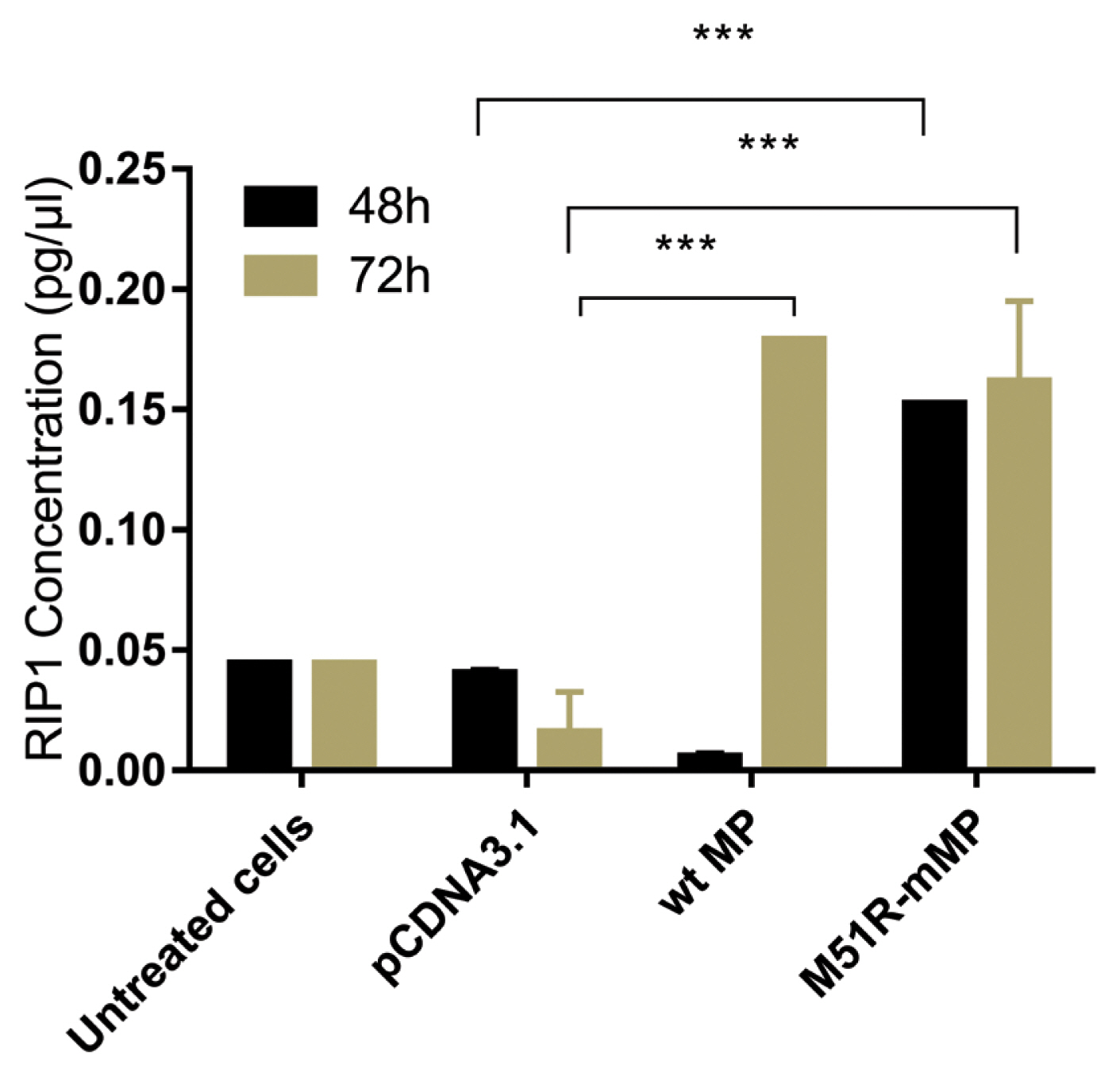
Figure 5
Induction of autophagic regulation in KYSE-30 cells transfected with wt and M51R-mMP.
Beclin1 was assayed in KYSE-30 cells that were untransfected or transfected with pcDNA3.1, wt, or M51R-mMP at 48 (black bars) and 72 (mustard bars) hours post-transfection. ** p < 0.001.
wt = wild-type; M51R-mMP = M51R-mutant matrix protein.
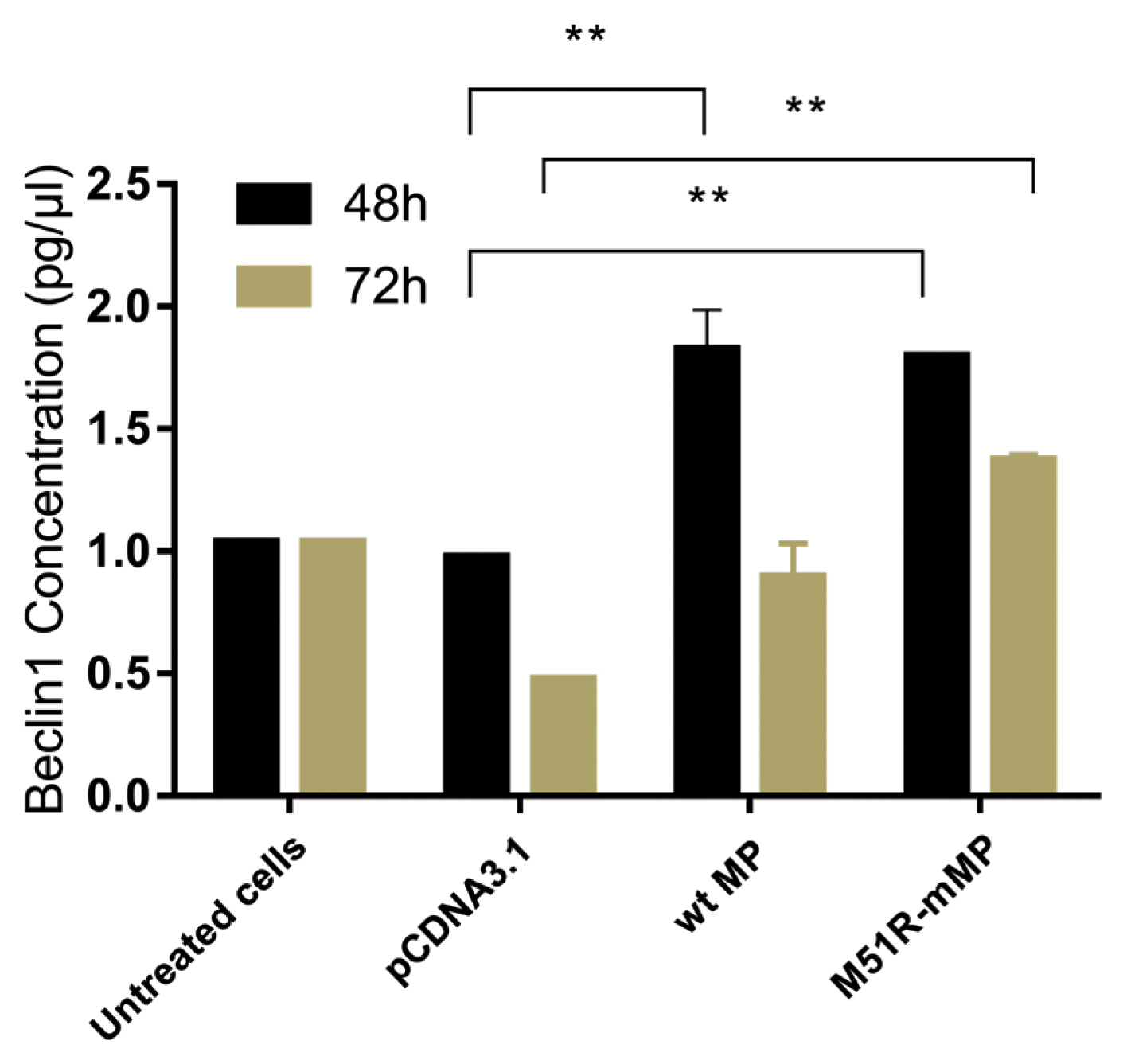
Figure & Data
References
Citations
Citations to this article as recorded by 
- Evoking pyroptosis with nanomaterials for cancer immunotherapy: Current boom and novel outlook
Wen-Da Wang, Zhi-Jun Sun
Nano TransMed.2022; 1(1): 9130001. CrossRef - Biological causes of immunogenic cancer cell death (ICD) and anti-tumor therapy; Combination of Oncolytic virus-based immunotherapy and CAR T-cell therapy for ICD induction
Amirhossein Mardi, Anastasia V. Shirokova, Rebar N. Mohammed, Ali Keshavarz, Angelina O. Zekiy, Lakshmi Thangavelu, Talar Ahmad Merza Mohamad, Faroogh Marofi, Navid Shomali, Amir Zamani, Morteza Akbari
Cancer Cell International.2022;[Epub] CrossRef - Oncolytic Viruses: Immunotherapy Drugs for Gastrointestinal Malignant Tumors
Qingbo Li, Patrick Kwabena Oduro, Rui Guo, Ruiqiao Li, Ling Leng, Xianbin Kong, Qilong Wang, Long Yang
Frontiers in Cellular and Infection Microbiology.2022;[Epub] CrossRef - Live-attenuated poliovirus-induced extrinsic apoptosis through Caspase 8 within breast cancer cell lines expressing CD155
Hossein Vazeh, Emad Behboudi, Anahita Hashemzadeh-Omran, Abdolvahab Moradi
Breast Cancer.2022; 29(5): 899. CrossRef - Exogenous expression of both matrix protein and glycoprotein facilitates infectious viral particle production of Borna disease virus 1
Takehiro Kanda, Madoka Sakai, Akiko Makino, Keizo Tomonaga
Journal of General Virology .2022;[Epub] CrossRef - La herencia de Prometeo. Las enfermedades ocupacionales en el Corpus Hippocraticum
César Sierra Martín
Asclepio.2022; 74(1): p587. CrossRef - Analyses of cell death mechanisms related to amino acid substitution at position 95 in the rabies virus matrix protein
Isshu Kojima, Fumiki Izumi, Makoto Ozawa, Yoshikazu Fujimoto, Misuzu Okajima, Naoto Ito, Makoto Sugiyama, Tatsunori Masatani
Journal of General Virology .2021;[Epub] CrossRef - The role of non-apoptotic cell death in the treatment and drug-resistance of digestive tumors
Yang Yang, LiangLiang Bai, Weiting Liao, Mingyang Feng, Mengxi Zhang, Qiuji Wu, Kexun Zhou, Feng Wen, Wanting Lei, Nan Zhang, Jiaxing Huang, Qiu Li
Experimental Cell Research.2021; 405(2): 112678. CrossRef - NEBL and AKT1 maybe new targets to eliminate the colorectal cancer cells resistance to oncolytic effect of vesicular stomatitis virus M-protein
Zoleikha Mamizadeh, Mohamad Reza Kalani, Masoud Parsania, Mohammad Mehdi Soltan Dallal, Abdolvahab Moradi
Molecular Therapy - Oncolytics.2021; 23: 593. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite




