Articles
- Page Path
- HOME > Osong Public Health Res Perspect > Volume 14(6); 2023 > Article
-
Original Article
Genetic diversity and evolutionary patterns of SARS-CoV-2 among the Bhutanese population during the pandemic -
Tshering Dorji
 , Kunzang Dorji, Tandin Wangchuk, Tshering Pelki, Sonam Gyeltshen
, Kunzang Dorji, Tandin Wangchuk, Tshering Pelki, Sonam Gyeltshen -
Osong Public Health and Research Perspectives 2023;14(6):494-507.
DOI: https://doi.org/10.24171/j.phrp.2023.0209
Published online: December 14, 2023
Royal Centre for Disease Control, Ministry of Health, Royal Government of Bhutan, Thimphu, Bhutan
- Corresponding author: Tshering Dorji Royal Centre for Disease Control, Wangchutaba, Serbithang, Thimphu 11001, Bhutan E-mail: cerorziks@gmail.com
© 2023 Korea Disease Control and Prevention Agency.
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
- 1,203 Views
- 49 Download
Abstract
-
Objectives
- The coronavirus disease 2019 (COVID-19) pandemic, caused by a dynamic virus, has had a profound global impact. Despite declining global COVID-19 cases and mortality rates, the emergence of new severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants remains a major concern. This study provides a comprehensive analysis of the genomic sequences of SARS-CoV-2 within the Bhutanese population during the pandemic. The primary aim was to elucidate the molecular epidemiology and evolutionary patterns of SARS-CoV-2 in Bhutan, with a particular focus on genetic variations and lineage dynamics.
-
Methods
- Whole-genome sequences of SARS-CoV-2 collected from Bhutan between May 2020 and February 2023 (n=135) were retrieved from the Global Initiative on Sharing All Influenza Database.
-
Results
- The SARS-CoV-2 variants in Bhutan were predominantly classified within the Nextstrain clade 20A (31.1%), followed by clade 21L (20%) and clade 22D (15.6%). We identified 26 Pangolin lineages with variations in their spatial and temporal distribution. Bayesian time-scaled phylogenetic analysis estimated the time to the most recent common ancestor as February 15, 2020, with a substitution rate of 0.97×10–3 substitutions per site per year. Notably, the spike glycoprotein displayed the highest mutation frequency among major viral proteins, with 116 distinct mutations, including D614G. The Bhutanese isolates also featured mutations such as E484K, K417N, and S477N in the spike protein, which have implications for altered viral properties.
-
Conclusion
- This is the first study to describe the genetic diversity of SARS-CoV-2 circulating in Bhutan during the pandemic, and this data can inform public health policies and strategies for preventing future outbreaks in Bhutan.
- The coronavirus disease 2019 (COVID-19) pandemic, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has had a profound global impact. It has resulted in a significant loss of human life and has posed unprecedented threats to public health systems, food security, economies, and education [1]. On May 5, 2023, the World Health Organization (WHO) declared that COVID-19 no longer constituted a Public Health Emergency of International Concern. Instead, it was recognized as an established and ongoing health challenge [2]. As of May 18, 2023, there have been over 766 million confirmed cases of COVID-19, leading to 6.9 million reported deaths worldwide. In Bhutan, there have been 62,668 confirmed cases of COVID-19, resulting in 21 deaths [3].
- The SARS-CoV-2 virus is subject to frequent mutations, which result in the emergence of various new variants. The complete genomic sequence of SARS-CoV-2 was first published on January 3, 2020 [4]. Since that time, over 151 thousand genomic sequences have been submitted to the Global Initiative on Sharing All Influenza Database (GISAID) [5]. The SARS-CoV-2 genome is composed of a positive-sense, single-stranded RNA of approximately 30,000 nucleotides, with a guanine-cytosine (GC) content of 38% [6]. It encodes and expresses a total of 12 proteins. The genome includes 13 to 15 open reading frames (ORFs), of which 12 are functional. Two-thirds of the genome is made up of 2 ORFs, ORF1a and ORF1b, which encode 2 polyproteins, pp1a and pp1ab, through a ribosomal frameshifting mechanism. These polyproteins are subsequently processed by viral proteases into 16 non-structural proteins (NSPs), which facilitate the replication and transcription of the viral RNA. The virus forms a membrane-associated replication and transcription complex that shields the viral RNA from the host’s innate immune sensors, thereby enhancing viral replication efficiency [7,8]. Notably, the ORF1ab and 4 structural proteins (spike, envelope, membrane, and nucleocapsid) are arranged in a 5′-3′ order and are potential targets for drugs and vaccines. In addition, the genome encodes accessory proteins and other NSPs via the ORF3a, ORF6, ORF7a, ORF7b, ORF8, and ORF10 genes [8].
- Nucleotide variations in genes can alter the structure and function of viral proteins. These mutations can influence viral characteristics, potentially triggering new outbreaks, reducing vaccine effectiveness, and impeding the development of new drugs and diagnostic kits [9]. Initially, genetic lineages were identified based on the phylogenetic framework of GISAID [5], Nextstrain [10], or Pangolin [11]. In response to the emergence of new variants and to avoid stigmatizing the countries where these key variants originated, the WHO established a working group of experts. This group is responsible for monitoring the virus's evolution and creating a nomenclature to differentiate these variants. The working group assigned these variants with letters from the Greek alphabet and classified them as either variants of interest (VOIs) or variants of concern (VOCs), depending on their transmissibility, severity, and ability to evade the immune system [12].
- While the overall number of COVID-19 cases is on the decline, thanks to increasing population immunity and decreasing mortality rates, the ongoing evolution of SARS-CoV-2 and the emergence of VOIs and VOCs remain major global concerns [13]. These variants, which include Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), Omicron BA.2 (B.1.1.529.2), BQ.1 (B.1.1.529.3), and XBB (B.1.1.529.5), have demonstrated increased transmissibility, immune evasion, or diminished vaccine efficacy in comparison to the original strain [14]. It is essential to monitor the circulation and impact of these variants in order to adjust public health measures as necessary.
- The primary objective of this study was to identify the variants and amino acid mutations of SARS-CoV-2 strains that circulated in Bhutan during the pandemic. We conducted a comparative analysis of the nucleotide and amino acid sequences of Bhutanese SARS-CoV-2 strains, using the Wuhan-Hu-1 strain as a reference. In addition, we explored the phylogenetic relationship among these strains to gain insight into their evolutionary links. Our focus was on pinpointing specific mutations and alterations in the amino acid sequence of functional proteins within the SARS-CoV-2 strains from Bhutan, including the spike, envelope, membrane, and nucleocapsid proteins. We analyzed samples collected from Bhutan over an extended period, from May 2020 to February 2023 (i.e., nearly 3 years), to track the evolutionary changes and trends of SARS-CoV-2 within the Bhutanese population. This extensive temporal distribution enabled us to observe changes over time in the virus's genetic composition and its potential influence on disease dynamics. This study represents the first report on the genomic characterization of SARS-CoV-2 in Bhutan.
Introduction
- Sequence Data Acquisition
- Full-genome sequences of SARS-CoV-2 from Bhutan were retrieved from the GISAID database (https://gisaid.org) for samples collected between May 2020 and February 2023. We only included high-quality whole-genome sequences that exceeded 29,000 base pairs (bp). All sequences were obtained in a multi-sequence FASTA format, and each was accompanied by metadata detailing patient status and sequencing technology. Our dataset consisted of 135 whole-genome sequences, distributed across the years as follows: 40 from 2020, 38 from 2021, 46 from 2022, and 11 from 2023. We downloaded the reference sequence NC_045512.2 as a separate FASTA file. Supplementary Material 1 includes the EPI_SET ID and the respective digital object identifier (DOI) for each SARS-CoV-2 sequence used in this study.
- Phylogenetic Analysis
- Nextstrain (https://nextstrain.org), an open-source bioinformatics visualization platform designed for the phylogenetic analysis of complete SARS-CoV-2 genomic sequences, was used. Nextclade ver. 2.13.0 [15], a web-based tool integrated within Nextstrain, was employed for sequence quality assessment and clade assignment. GISAID clades and WHO labels were determined using the GISAID CoVsurver tool (https://gisaid.org/) [16] and the coronavirus typing tool [17], respectively. The Pangolin webserver data ver. 1.19 (https://pangolin.cog-uk.io/) was used to further classify lineage typology. The geographical distribution of clades was visualized using Microreact (https://microreact.org).
- Sequence Alignment and Recombination Analysis
- To align the sequences, we obtained pairwise sequence alignment data in FASTA format from Nextclade and conducted multiple sequence alignment (MSA) using MAFFT ver. 7 (https://mafft.cbrc.jp/alignment/software/) [18]. This ensured optimal sequence congruence for the following procedures. To explore the presence of recombination events and pinpoint breakpoints within the aligned sequences, we utilized the Recombinant Detection Program ver. 4.5 (RDP4, http://web.cbio.uct.ac.za/~darren/rdp.html) [19]. This program incorporates various algorithms, including RDP, GENECONV, Maxchi, Chimaera, SiSscan, PhylPro, and 3Seq. We constructed a maximum likelihood phylogenetic tree directly using RDP4, in conjunction with the RAxML8 [20] algorithm, which takes recombination events into consideration. The final tree annotation, which depicts the evolutionary relationships among the SARS-CoV-2 sequences, was produced using iTOL ver. 6.7.4 (https://itol.embl.de/) [21].
- Estimation of Time to the Most Recent Common Ancestor
- To calculate the time to the most recent common ancestor (TMRCA) and the 95% highest posterior density (HPD) of SARS-CoV-2 genomes from Bhutan, we performed a Bayesian Markov chain Monte Carlo (MCMC) [22] analysis using BEAST ver. 1.10.4 (https://beast.community/) [23]. The MSA created by MAFFT was imported into BEAST. We used a Hasegawa-Kishino-Yano substitution model [24] for nucleotide frequencies and kappa, along with a strict molecular clock with uniform rates across branches (rate set to 1.0). We also used a coalescent exponential growth model with a Laplace prior (scale 100), and a coalescent population size prior distribution uniform between 0 and 10. The analysis started with a random starting tree. We ran a total of 100 million MCMC steps, sampling and logging parameters every 10,000 generations. We verified the effective sample sizes using Tracer ver. 1.7.2 (https://beast.community/tracer) [25] to ensure a sample size greater than 2,000. We extracted the TMRCA and the 95% HPD from the BEAST output log files using Tracer ver. 1.7.2. The maximum clade credibility (MCC) tree was generated using TreeAnnotator ver. 1.4 (https://beast.community/treeannotator), discarding the first 10% as burn-in. Finally, we visualized the final tree using FigTree v1.4 (http://tree.bio.ed.ac.uk/software/figtree/).
- Mutation Analysis
- Both nucleotide and amino acid mutations were identified with the SARS-CoV-2 isolate Wuhan-Hu-1 (NC_045512.2) as a reference, using the GISAID CoVsurver tool and the coronavirus typing tool. We analyzed the frequency of mutations for all 135 sequences, and the frequency of mutations for each protein was calculated based on the total number of samples with point mutations.
- IRB Approval
- Ethical clearance and approval were not applicable since the data used in this study was obtained from a publicly available database, GISAID. No additional demographic or clinical information was used in this study.
Materials and Methods
- SARS-CoV-2 Genome Analysis
- We conducted a comprehensive analysis of the genetic diversity in 135 SARS-CoV-2 genomes, which were gathered from Bhutan between May 2020 and February 2023. These genomes were classified using various nomenclature systems, such as GISAID, Nextstrain, WHO, and Pangolin, through phylogenetic analyses. Furthermore, we offered in-depth insights into the demographic characteristics of the sample set, the quality of the genomes, and their temporal distribution.
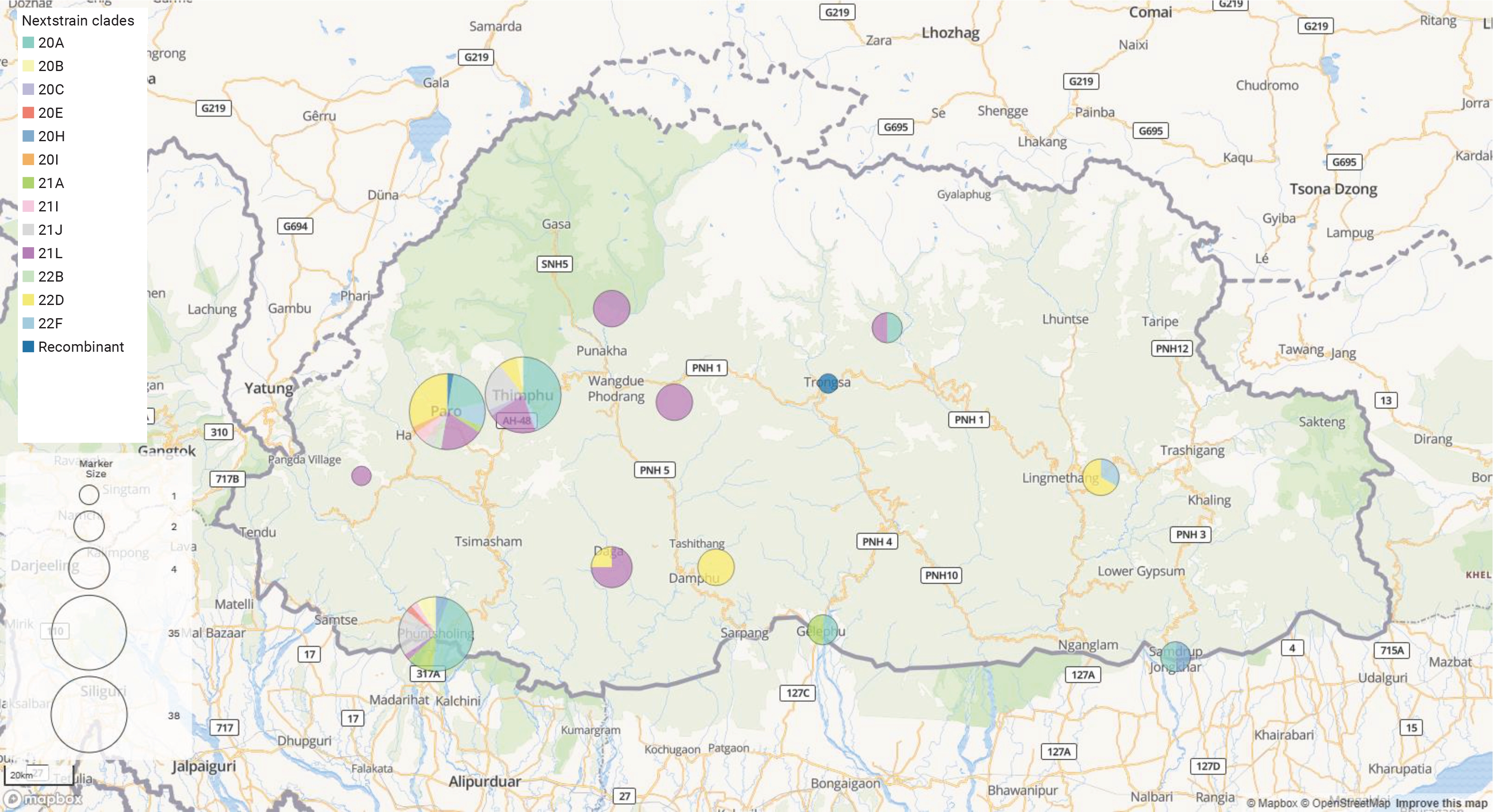
- The distribution of the SARS-CoV-2 genome across Bhutan was as follows: both Paro and Thimphu had 38 genomes each, while Phuentsholing had 35 genomes. Other locations represented in the dataset included Dagana with 4 genomes, and Punakha, Tsirang, and Mongar, each with 3 genomes. Furthermore, both Samdrupjongkhar and Gelephu had 2 genomes each, while Trongsa and Haa each had a single genome. Our sample set comprised 80 males and 55 females, with ages ranging from 10 to 85 years and a median age of 36 years. The average length of the viral genome was 29,800 bp, with an average genome coverage of 98.8%. The average length of amino acids was found to be 9,691. According to Nextclade’s overall quality scoring system, of the 135 sequences analyzed, 131 were classified as good quality, while 4 were deemed mediocre quality. None of the sequences were flagged as bad quality.
- Phylogenetic Relationships among Bhutanese SARS-CoV-2 Viral Strains
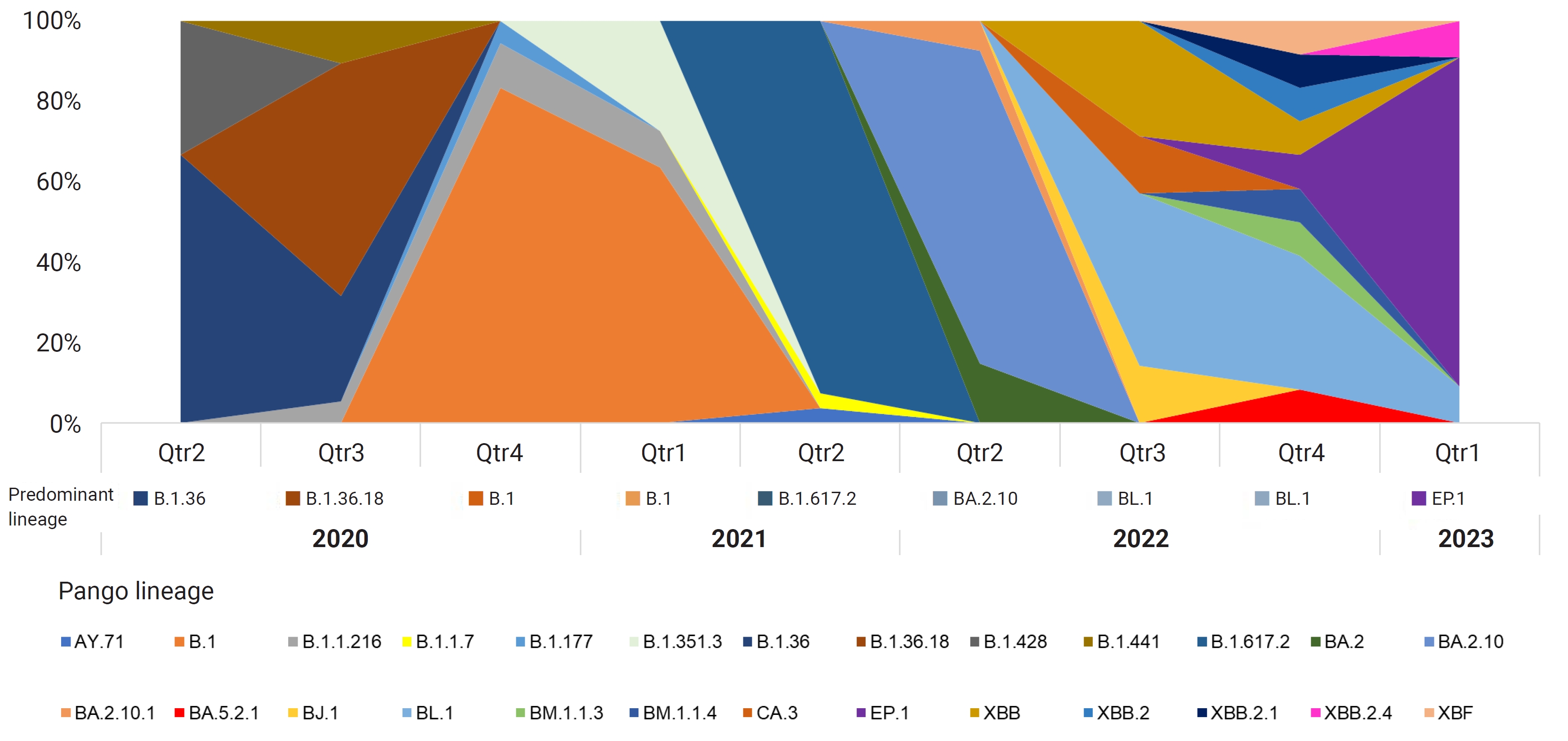
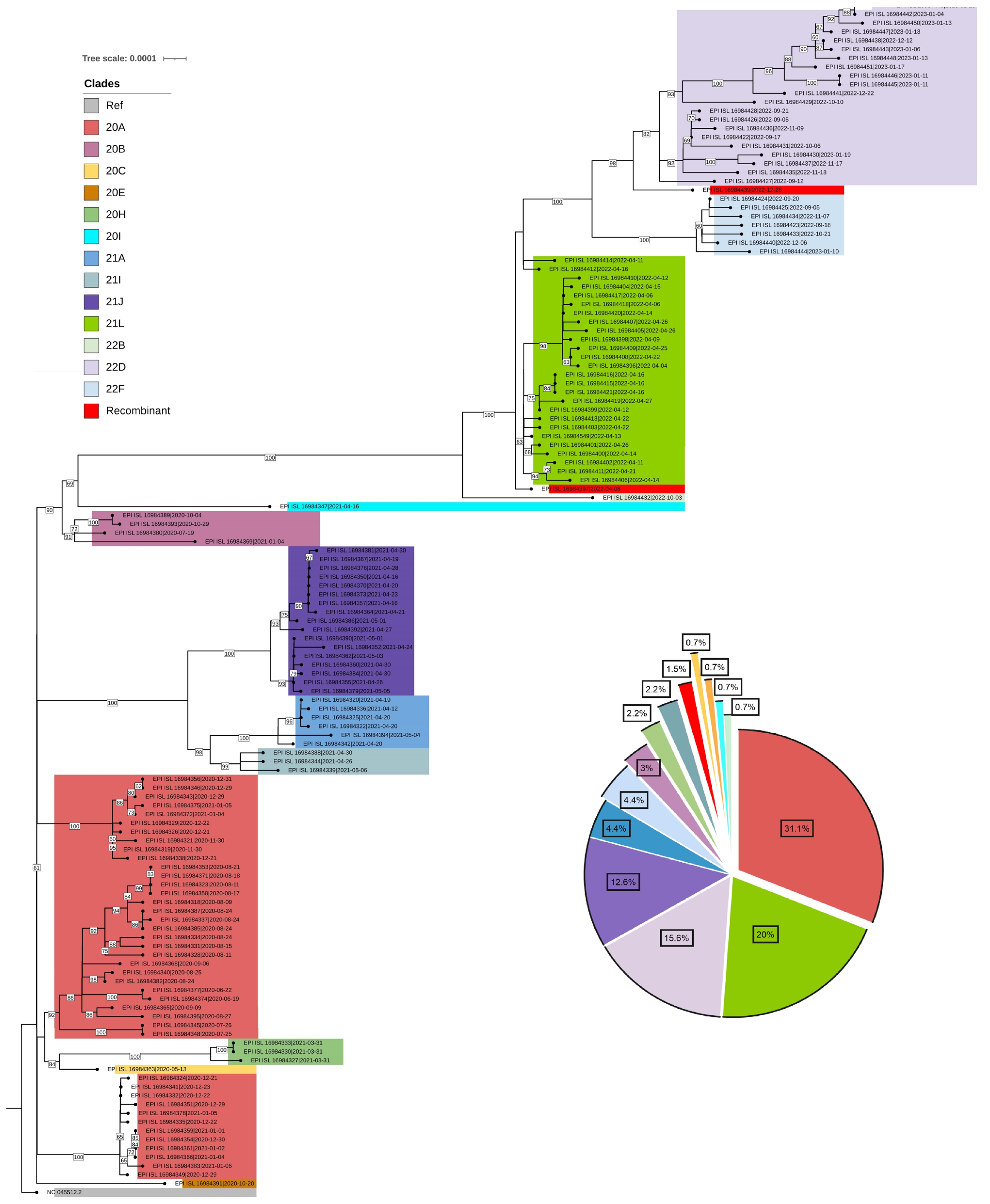
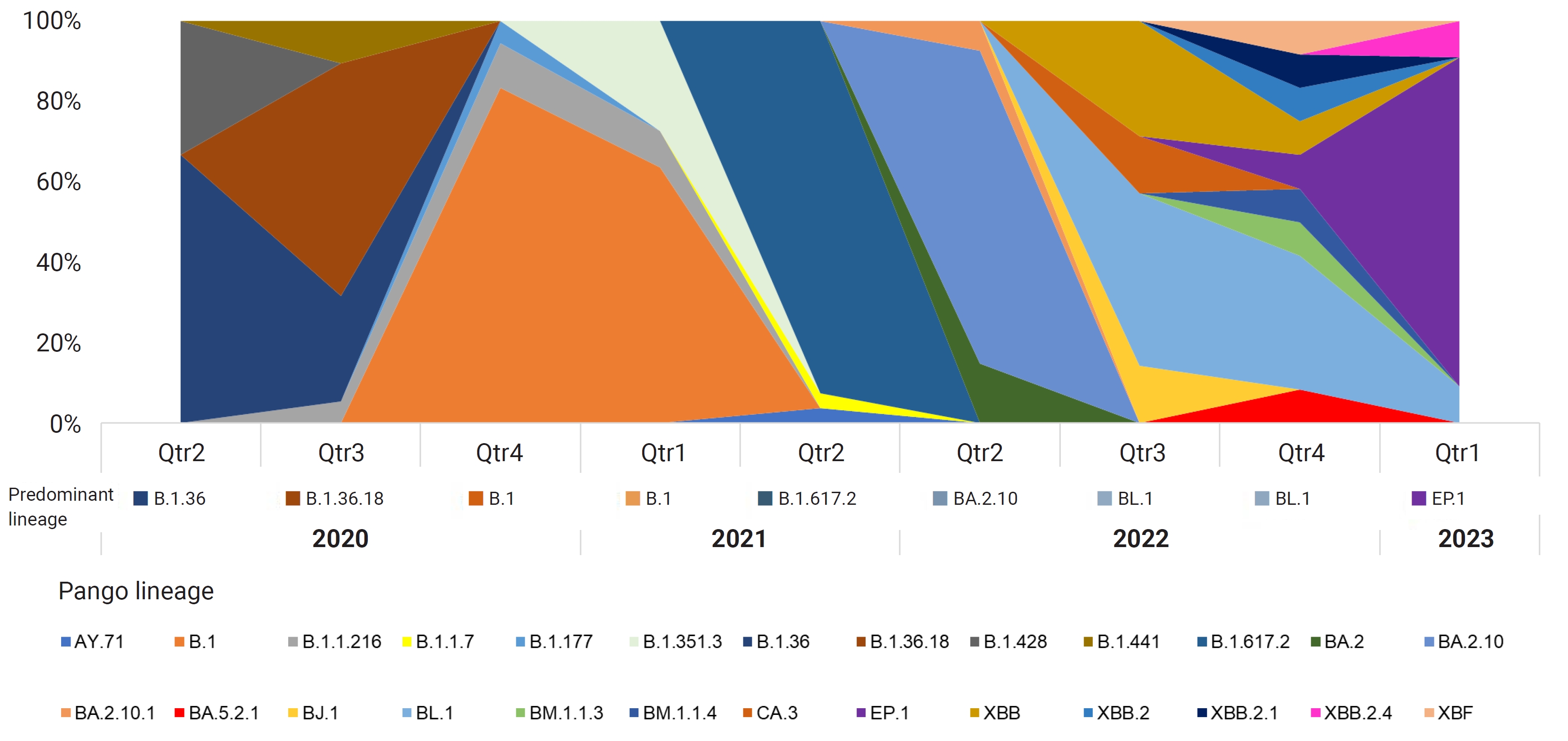
- After conducting a phylogenetic analysis, we identified 6 distinct GISAID clades (G, GH, GK, GR, GRA, and GV) to which the Bhutanese SARS-CoV-2 strains were assigned. The GH clade was predominant in 2020, succeeded by the G clade in 2021, and then the GRA clade in 2022 and early 2023. We also identified 4 distinct WHO labels within the Bhutanese SARS-CoV-2 strains: Alpha, Beta, Delta, and Omicron. Notably, approximately 35.6% of the sequences did not match any WHO label and were instead assigned to international A_B diversity by the coronavirus typing tool. From May 2020 to February 2023, we detected a total of 29 Pangolin lineages among the strains. The B.1 variant was the most prevalent in 2020, accounting for 37.7% of the cases. In 2021, the dominant variant was B.1.617, representing 65% of the cases. By 2022, the BA.2.10 lineage became prominent, later transitioning to EP.1, which accounted for 81.8% of the cases in early 2023. The distribution of Pangolin lineages within the samples showed temporal variations, reflecting the dynamic nature of the viral strains circulating in Bhutan (Figure 1). Table 1 provides a detailed frequency distribution of each nomenclature system. We also identified 2 recombinant variants, classified as Pangolin lineages BA.2 and XBF, using Nextclade and the coronavirus typing tool. To further explore recombination events, we employed RDP ver. 4.5 software, which confirmed the occurrence of recombination in at least 3 Bhutanese SARS-CoV-2 strains. These findings strongly suggest genetic recombination, highlighting the significant genetic diversity of SARS-CoV-2 in Bhutan. Detailed information on the recombination events in Bhutanese SARS-CoV-2 genomes can be found in Supplementary Material 2.
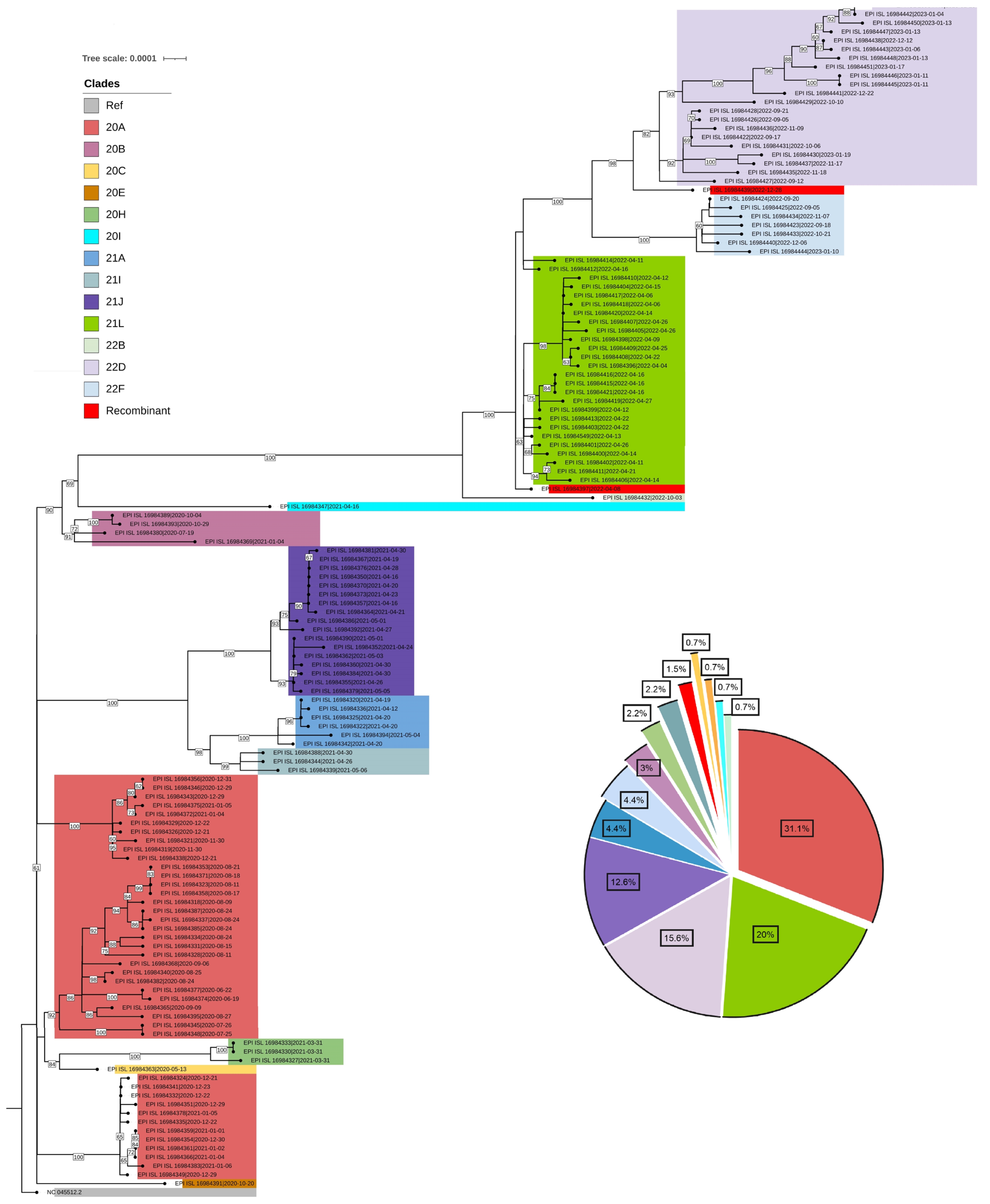
- To infer the origin and evaluate potential divergence events of SARS-CoV-2, we created a maximum likelihood phylogenetic tree. This was based on 135 complete genomes from isolates in Bhutan and the reference virus from Wuhan, China (NC_045512.2), using RDP4 (with RAxML8). The tree demonstrated a swift diversification of SARS-CoV-2, spread across 14 distinct Nextstrain clades. These included 20A, 20B, 20C, 20E, 20H, 20I, 21A, 21I, 21J, 21L, 22B, 22D, and 22F (Figures 2, 3).
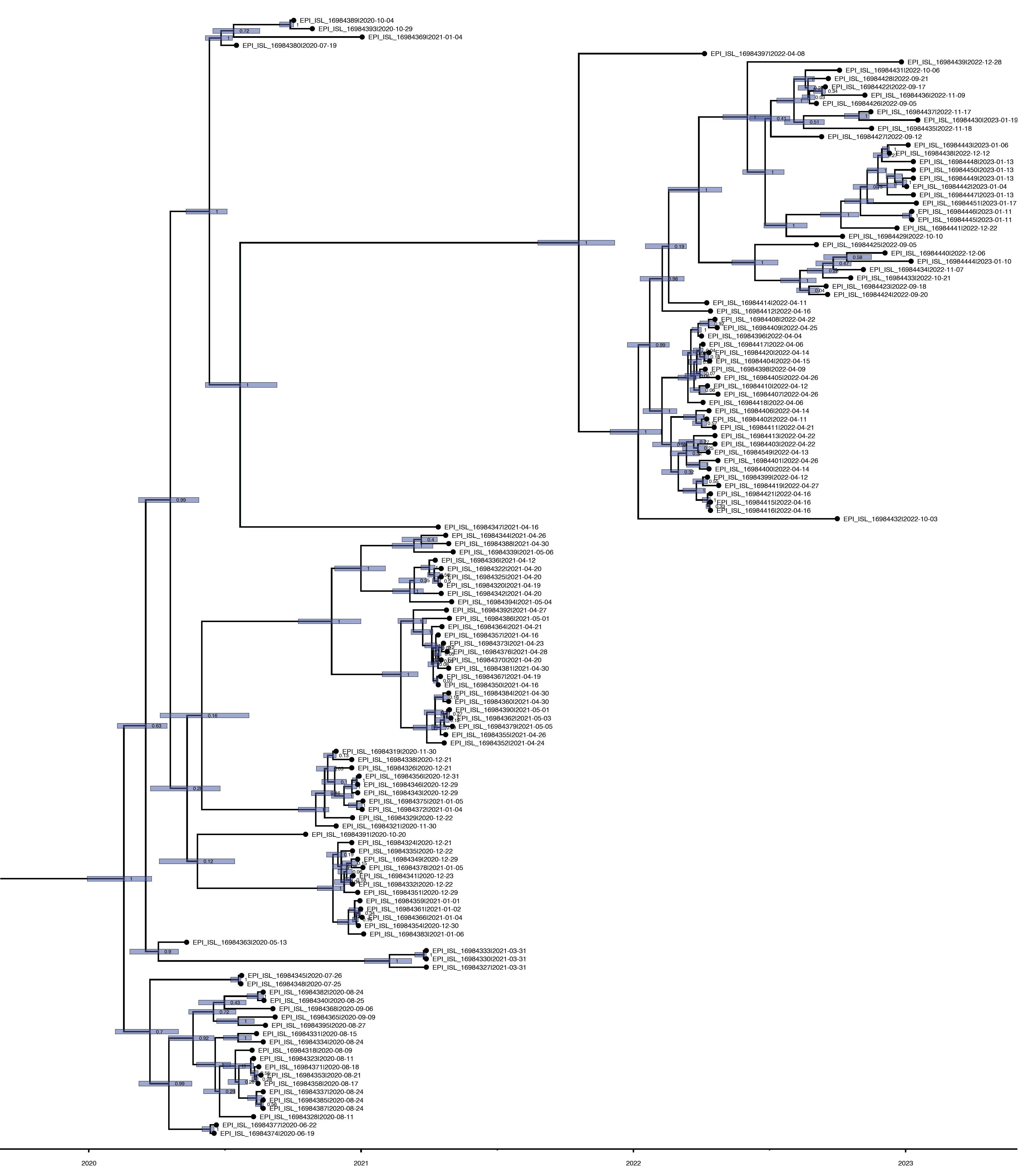
- Time to the Most Recent Common Ancestor of SARS-CoV-2 in Bhutan
- The TMRCA for the SARS-CoV-2 strains circulating in Bhutan was estimated to be around February 15, 2020, with a 95% HPD interval ranging from January 3, 2020 to September 13, 2020. The mean substitution rate was calculated to be 0.97×10–3 substitutions per site per year, with a 95% HPD of 0.85×10–3 to 1.10×10–3. Figure 4 illustrates the MCC phylogenetic relationships among the SARS-CoV-2 genomes, as determined from the Bayesian coalescent framework using the coalescent exponential growth and tip-dating method.
- Mutation Analysis
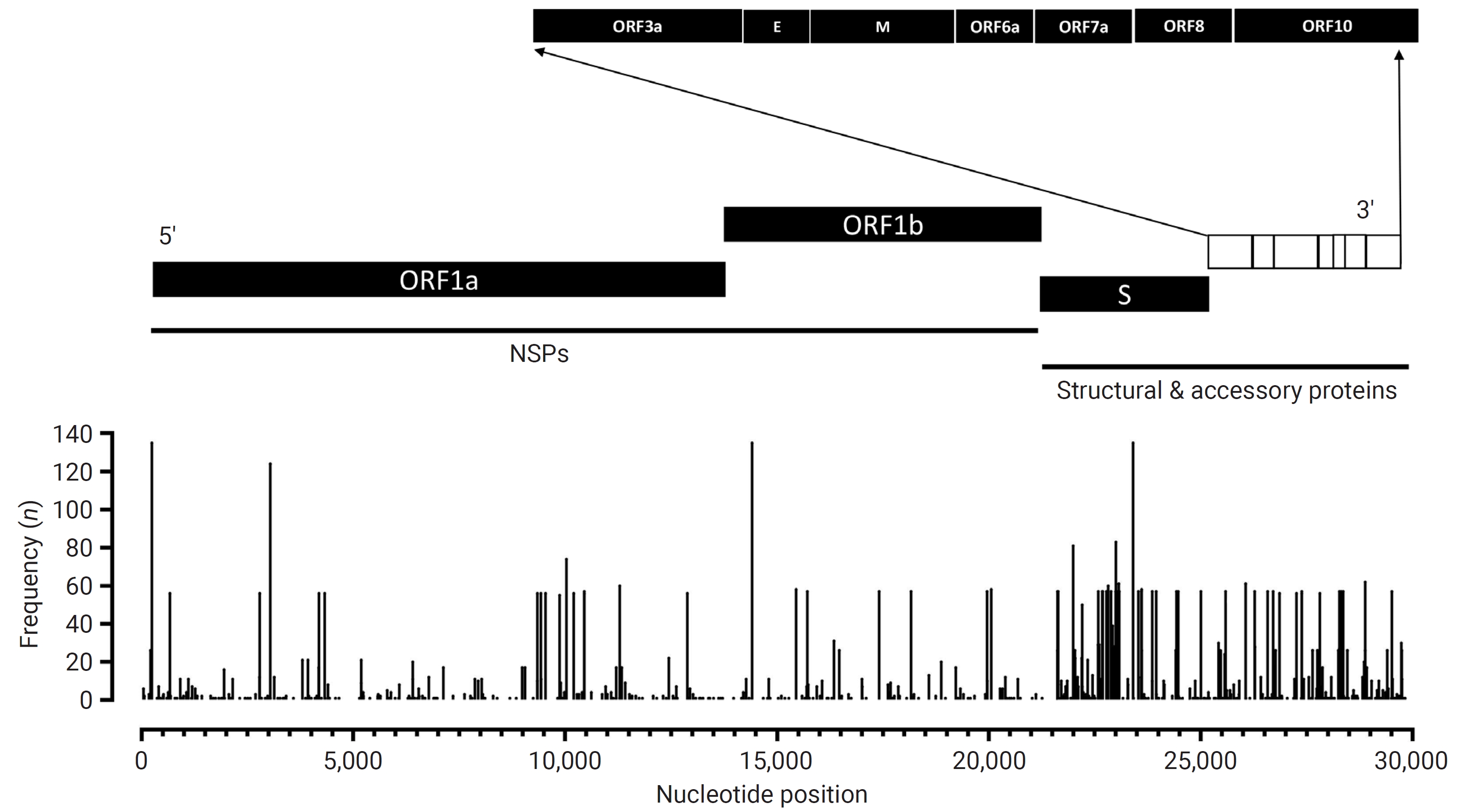
- The mutational revealed 9,413 single-nucleotide variations (SNVs) within the SARS-CoV-2 genomes. These SNVs comprised 6,568 substitutions and 2,845 deletions. As a result of these SNVs, 5,613 amino acid changes were observed, which included 4,762 amino acid substitutions and 851 deletions. Figure 5 illustrates the locations and frequencies of the nucleotide variations plotted along the genomic sequence of the reference SARS-CoV-2 (NC_045512.2). Among the SNVs, the most common alteration was C>T, which accounted for 40% of the observed SNVs, followed by G>A, making up 12%. Importantly, no large deletions or insertions exceeding 50 nucleotides were detected. The longest deletion identified extended from position 29734 to 29759, resulting in the removal of a 26-nucleotide sequence (GAGGCCACGCGGAGTACGATCGAGTG).
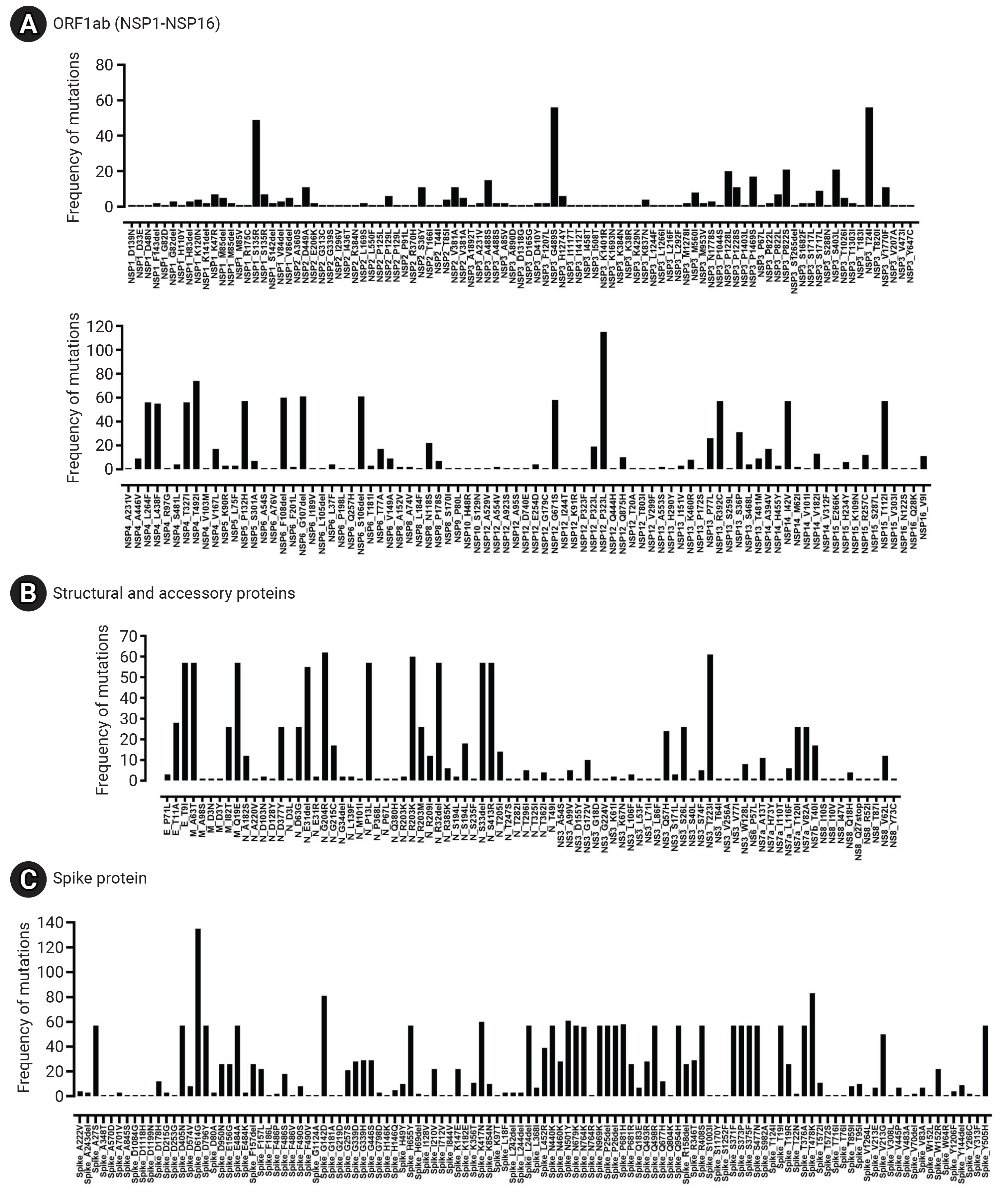
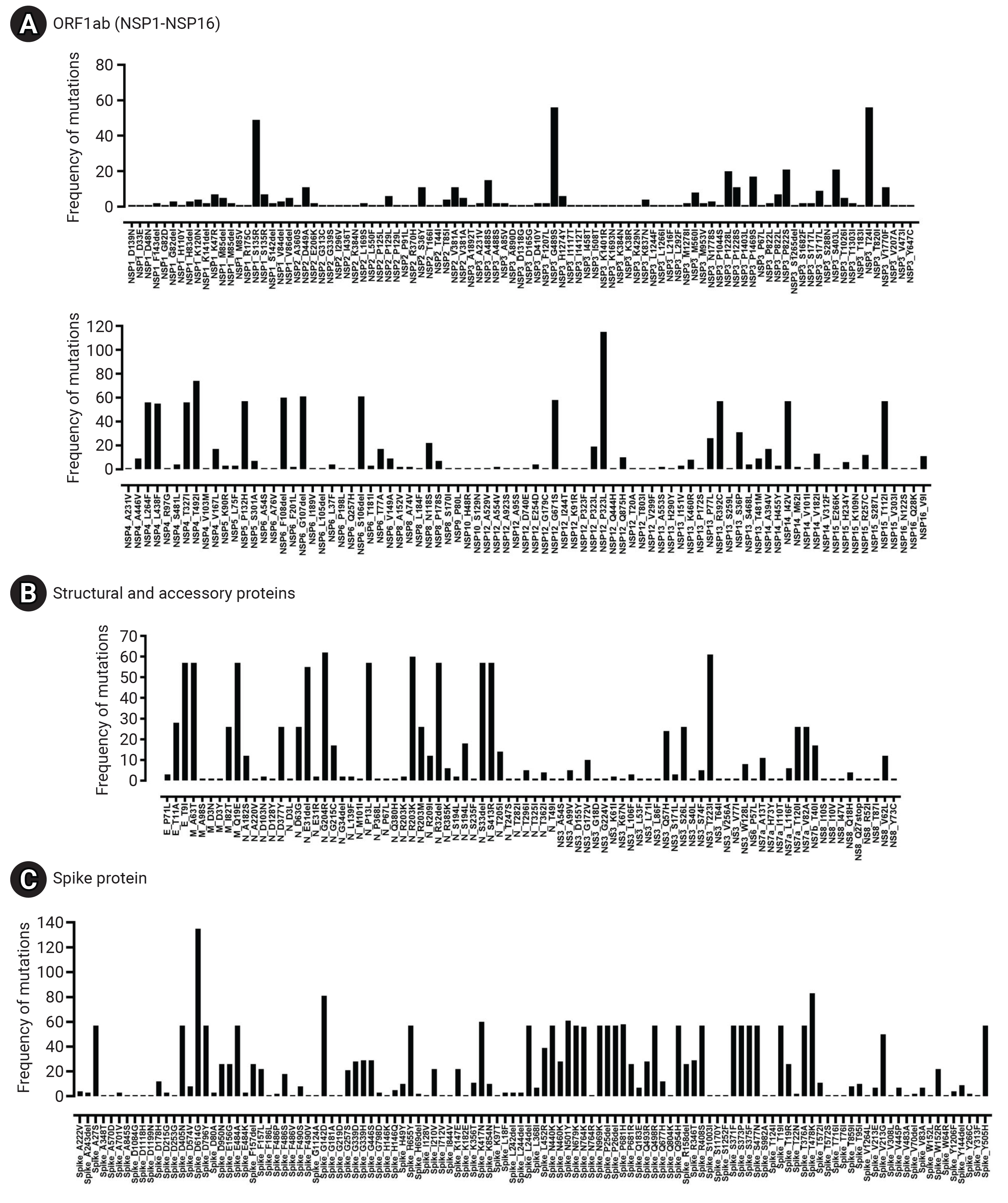
- Frequency of Mutations among Major Viral Proteins
- The spike glycoprotein (S) displayed the highest mutation frequency among the major viral proteins, with 116 unique types identified. The most common amino acid substitutions in S were D614G (135/135), T478K (83/135), G142D (81/135), and N501Y (61/135). In contrast, NSP7 and NSP11 were highly conserved, with no mutations detected. NSP10 (2/135), NSP9 (1/135), NS6 (1/135), and NS7a (1/135) also exhibited low mutation rates. The specific frequency of amino acid mutations in the major proteins of Bhutanese SARS-CoV-2 viral strains is illustrated in Figure 6 and Table 2.
Results
Single-nucleotide variations
- In this study, we conducted a comprehensive analysis of 135 whole-genome sequences of SARS-CoV-2, collected from Bhutan between May 2020 and February 2023. These sequences, obtained from the GISAID database, were compared with the Wuhan-Hu-1 (NC_045512) reference strain. We characterized the genomic variation, as well as the phylogenetic and evolutionary dynamics, of SARS-CoV-2 from Bhutan. Our phylogenetic analysis revealed that multiple SARS-CoV-2 lineages were circulating in Bhutan in 2020, 2021, 2022, and early 2023. We discovered that the virus in Bhutan was distributed across 14 Nextstrain clades and 26 Pangolin lineages. In 2020, the predominant variant was B.1, accounting for 37.7% of the sequenced samples. This variant, which exhibited a global distribution in 2020, gave rise to more than 70 sub-lineages, indicating its extensive diversification [11]. In 2021, the dominant lineage in Bhutan shifted to B.1.617, prevalent in 65% of the sequenced samples. This variant, B.1.617, carried 3 key mutations (L452R, E484Q, and P681R) in the viral spike protein and was first detected in India in December 2020 [26]. Lineage B.1.617.2, a sub-lineage of B.1.617 also known as the Delta variant, gradually replaced the B.1.1.7 variant as the dominant global strain by March 2021 [27].
- In 2022, the most prevalent lineage of SARS-CoV-2 in Bhutan was BA.2.10 (Omicron), accounting for 48% of the sequenced samples. This variant first appeared in South Africa in November 2021 and led to a worldwide surge in COVID-19 cases due to its increased transmissibility compared to BA.1 [28]. Bhutan also experienced a significant increase in COVID-19 cases that year, resulting in a series of lockdowns and widespread testing [29]. Interestingly, a similar trend was observed in North India, where the Omicron BA.2 lineage became prominent during the third wave of COVID-19 cases, as documented by Zaman et al. [30]. This concurrent emergence of the Omicron variant in both Bhutan and North India prompts inquiries and hypotheses about potential links between these events and the dynamics of the cross-border transmission of the variant.
- The mutations observed in Bhutanese strains offer significant insights into the country's viral dynamics and their potential impact on global trends. Like many other regions, Bhutan has seen dynamic changes in the dominant SARS-CoV-2 lineages throughout the study period. This mirrors global trends and highlights the role of international travel and viral transmission networks. It further emphasizes the interconnectedness of Bhutan’s SARS-CoV-2 dynamics with the wider global landscape.
- SARS-CoV-2 exhibits a high rate of genomic mutations, and our study identified both SNVs and the distribution of point mutations across the major viral proteins. The most frequently detected SNVs in SARS-CoV-2 genomes from Bhutan were C>T, followed by G>A. This aligns with previous studies, which have reported similar findings and suggested a potential explanation. This explanation posits that there is a selective mutation pressure to decrease the number of CpG sites in the virus’s genome. This is due to the abundance of human antiviral proteins, such as APOBEC3 and ZAP [31,32]. Among the proteins we analyzed, the spike glycoprotein exhibited the highest number of point mutations. This was followed by N, NSP3, NSP12, and NSP4, in that order. These proteins play crucial roles in the viral life cycle and are promising targets for vaccines and therapeutic interventions [33]. The high frequency of point mutations in these proteins raises questions about the effectiveness of drug candidates being investigated as potential therapies or diagnostics that target these proteins. High mutation rates can undermine the efficacy of targeted treatments, potentially weakening their ability to disrupt viral replication or inhibit viral protein function.
- One of the key findings of our study is the abundance of mutations in the spike glycoprotein, a vital component in viral infection and pathogenesis. We observed several noteworthy mutations in the spike protein of Bhutanese strains, including D614G, T478K, G142D, N501Y, K417N, P681H, A27S, D405N, D796Y, E484A, H655Y, L24del, N440K, N679K, N969K, P25del, P26del, Q498R, Q954H, R408S, S371F, S373P, S375F, S477N, T19I, T376A, Y505H, N764K, and V213G. These mutations were identified in over 50 samples. Among the functional proteins of the SARS-CoV-2 virus, the spike protein shows the highest mutation rate. Many studies have underscored the significance of mutated residues on the S-receptor binding domain (S-RBD) of the Omicron variant, as they enhance its binding affinity with ACE2 [34].
- The mutations observed in Bhutanese strains are of significant importance due to their potential impact on various aspects of the virus's behavior. For instance, the D614G mutation, which is commonly observed, has been reported in 99.03% of samples from 212 countries worldwide [5]. Strains carrying this mutation have been found to display lower reverse transcription-polymerase chain reaction cycle thresholds, higher upper respiratory tract viral loads without an increase in disease severity, and enhanced infectivity [14]. Another mutation, N501Y, found in Bhutanese strains, is known to modify the shape of the RBD, thereby increasing its binding affinity with human ACE2 receptors. This mutation is present in VOCs, such as those first identified in the UK (B.1.1.7), South Africa (B.1.351), and Brazil (P.1) [35,36]. These variants have been demonstrated to spread more rapidly than other lineages and may also affect vaccine efficacy to some extent. However, there is no evidence to suggest that they cause more severe disease or increase the risk of death. Other mutations, such as E484K, K417N, and S477N in the spike protein, have also been associated with changes in viral properties [37] and were frequently found among strains from Bhutan. These findings highlight the potential implications of these mutations for viral pathogenesis, transmission, and vaccine effectiveness. Further research is required to gain a better understanding of their specific roles in disease dynamics.
- In addition to the mutations in the spike protein, our analysis also investigated mutations in other key viral proteins, providing insights into potential therapeutic targets and diagnostic challenges. The discovery of mutations in N, NSP3, NSP12, and NSP4 is especially significant, as these proteins play crucial roles in the viral life cycle and are promising targets for vaccine development and therapeutic interventions. However, the high mutation rates observed in these proteins raise questions about the efficacy of drug candidates being considered for potential therapies or diagnostics. The ever-changing nature of the virus highlights the need for ongoing research and development of antiviral therapies that can effectively combat these mutations.
- This study also analyzed non-mutated proteins, such as NSP7 and NSP11, emphasizing their conservation. Proteins that are highly conserved, including NSP10, NSP9, NS6, and NS7a, have crucial roles in the viral life cycle and interactions with the host. Previous studies have identified NSP10, NSP7, NSP8, NSP11, NSP16, ORF6, and NSP9 as highly conserved proteins in SARS-CoV-2 [38,39]. Mutations in these proteins could potentially impair their functions and decrease viral fitness. For example, NSP7 and NSP11 are co-factors of the viral polymerase NSP12 and are vital for its activity [38]. NSP10 enhances the processivity and fidelity of NSP12, acting as its co-factor [40]. NSP9, an RNA-binding protein, may aid in viral RNA synthesis. NS6 and NSP7a are accessory proteins that adjust the host immune response and disrupt interferon signaling [7]. The preservation of these proteins underscores their significance in sustaining viral functions. Approaches that target conserved regions or utilize combination therapies may offer potential solutions to the problems created by the virus’s mutational landscape, thereby enhancing the effectiveness of therapeutic interventions.
- The TMRCA of SARS-CoV-2 strains circulating in Bhutan was estimated in this study to have occurred around February 15, 2020, with a 95% HPD interval ranging from January 3, 2020 to September 13, 2020. This estimated TMRCA aligns with the timing of Bhutan’s first confirmed COVID-19 case, which was reported on March 5, 2020 [41]. Additionally, we calculated a substitution rate of 0.97×10–3 substitutions per site per year, consistent with findings from other studies [42–45].
- In response to the initial detection of its first COVID-19 case, Bhutan implemented swift and comprehensive measures, including the closure of all international borders, to prevent the local transmission of SARS-CoV-2. Of particular note is the remarkable success of Bhutan’s vaccination efforts, which underscores the effectiveness of the country’s vaccination strategy and the readiness of its citizens to accept the vaccine. Bhutan procured and administered vaccines through a mix of bilateral and multilateral donations from countries such as India, the United States, Denmark, China, Croatia, among others. By April 9, 2021, a total of 472,139 individuals (consisting of 250,362 men and 221,777 women) had been successfully vaccinated with the first dose of COVISHIELD (Oxford-AstraZeneca, produced by the Serum Institute of India), achieving an impressive coverage of 94% among the eligible population [46]. This success can be credited to effective mass communication and social engagement initiatives led by religious figures, De-Suup volunteers, and local leaders. These efforts were instrumental in dispelling myths and misinformation about the vaccine and motivating people to get vaccinated [47].
- The high rate of vaccination in Bhutan has not only significantly decreased the risk of severe illness, but it has also maintained COVID-19 morbidity at impressively low levels compared to neighboring countries in the region, such as India, Nepal, Bangladesh, and China. Throughout the entire pandemic, Bhutan has only reported 21 deaths attributed to SARS-CoV-2 [41]. This highlights the effectiveness of Bhutan’s proactive and well-organized public health response in protecting the health and well-being of its citizens.
Discussion
- This study offers a comprehensive overview of the genomic variation and evolutionary dynamics of SARS-CoV-2 in Bhutan throughout the course of the pandemic. We found that the SARS-CoV-2 strains present in Bhutan share common mutations with strains from other countries, but they are not identical. Our research revealed that multiple lineages have circulated in Bhutan and that the virus has adapted to environmental and selective pressures. These discoveries illuminate the transmission patterns, disease severity, and vaccine efficacy of SARS-CoV-2. The emergence of new variants with enhanced transmissibility and potential for immune evasion underscores the necessity for ongoing genomic surveillance and effective public health strategies, such as vaccine development and implementation. This study underscores the importance of genomic data in shaping the design and assessment of preventive and control measures against SARS-CoV-2 and its variants. Despite a decrease in overall cases, the virus continues to evolve, necessitating continued vigilance in our efforts to control the disease.
- Limitations
- A potential limitation of this study is sample representativeness, as it only included 135 whole-genome sequences of SARS-CoV-2 obtained from Bhutan between May 2020 and February 2023. This sample size is relatively small and may not accurately reflect the genetic diversity and distribution of SARS-CoV-2 throughout the country. Additionally, the dynamic evolution of SARS-CoV-2 could have given rise to new variants that were not detected or analyzed in this study, potentially impacting the relevance of the findings. Moreover, this study did not take into account other factors that could influence the transmission and outcome of SARS-CoV-2 infections.
Conclusion
- • A phylogenetic analysis of severe acute respiratory syndrome coronavirus 2 genomes from Bhutan revealed rapid diversification across 14 Nextstrain clades and 29 Pangolin lineages with temporal variations.
- • The time to the most recent common ancestor of the Bhutanese sequences was February 15, 2020 (95% highest posterior density [HPD], January 3, 2020 to September 13, 2020), with a substitution rate of 0.97×10–3 substitutions per site per year (95% HPD, 0.85×10–3 to 1.10×10–3).
- • The spike glycoprotein (S) exhibited the highest frequency of mutations among major viral proteins, with 116 distinct types, while NSP7 and NSP11 were highly conserved, with no mutations observed.
HIGHLIGHTS
Supplementary Material
Supplementary Material 1.
Supplementary Material 2.
-
Ethics Approval
Ethical clearance and approval were not applicable since the data used in this study were obtained from publicly available database, the Global Initiative on Sharing All Influenza Data (GISAID).
-
Conflicts of Interest
The authors have no conflicts of interest to declare.
-
Funding
None.
-
Availability of Data
The findings of this study are based on metadata associated with 135 sequences available on GISAID, and accessible at DOI: https://doi.org/10.55876/gis8.230622fy
-
Authors’ Contributions
Conceptualization: all authors; Data curation: all authors; Formal analysis: TD; Investigation: all authors; Methodology: all authors; Resources: all authors; Software: TD; Validation: all authors; Visualization: all authors; Writing–original draft: TD; Writing–review & editing: all authors. All authors read and approved the final manuscript.
Article information
The table shows the variants of SARS-CoV-2 from 135 Bhutanese sequences. The Pangolin variants are grouped according to 3 nomenclature systems (WHO, GISAID, and Nextstrain clades) along with the Pangolin lineages.
SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; WHO, World Health Organization; GISAID, Global Initiative on Sharing All Influenza Database.
SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; ORF, open reading frame; NSP, non-structural protein.
The proteins are grouped into structural and accessory proteins, ORF1a, and ORF1b. An amino acid change is a substitution, insertion, or deletion of 1 or more amino acids in the protein sequence. A mutation type is a classification of amino acid changes based on their effect on the protein structure and function. The table indicates the total number of amino acid changes and mutation types observed in each protein across all the sequenced samples.
- 1. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 2020;5:536−44.ArticlePubMedPMCPDF
- 2. World Health Organization (WHO). Statement on the fifteenth meeting of the IHR (2005) Emergency Committee on the COVID-19 pandemic [Internet]. WHO; 2023 [cited 2023 May 18]. Available from: https://www.who.int/news/item/05-05-2023-statement-on-the-fifteenth-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-coronavirus-disease-(covid-19)-pandemic.
- 3. World Health Organization (WHO). WHO coronavirus (COVID-19) dashboard [Internet]. WHO; 2023 [cited 2023 May 18]. Available from: https://covid19.who.int/.
- 4. Tan W, Zhao X, Ma X, et al. A novel coronavirus genome identified in a cluster of pneumonia cases-Wuhan, China 2019-2020. China CDC Wkly 2020;2:61−2.ArticlePubMedPMC
- 5. Elbe S, Buckland-Merrett G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob Chall 2017;1:33−46.ArticlePubMedPMCPDF
- 6. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020;579:265−9.ArticlePubMedPMCPDF
- 7. Gorkhali R, Koirala P, Rijal S, et al. Structure and function of major SARS-CoV-2 and SARS-CoV proteins. Bioinform Biol Insights 2021;15:11779322211025876. ArticlePubMedPMCPDF
- 8. Naqvi AA, Fatima K, Mohammad T, et al. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim Biophys Acta Mol Basis Dis 2020;1866:165878. ArticlePubMedPMC
- 9. Wu S, Tian C, Liu P, et al. Effects of SARS-CoV-2 mutations on protein structures and intraviral protein-protein interactions. J Med Virol 2021;93:2132−40.ArticlePubMedPMCPDF
- 10. Hadfield J, Megill C, Bell SM, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 2018;34:4121−3.ArticlePubMedPMCPDF
- 11. Rambaut A, Holmes EC, O'Toole A, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol 2020;5:1403−7.ArticlePubMedPMCPDF
- 12. Konings F, Perkins MD, Kuhn JH, et al. SARS-CoV-2 variants of interest and concern naming scheme conducive for global discourse. Nat Microbiol 2021;6:821−3.ArticlePubMedPDF
- 13. Liu S, Jia Z, Nie J, et al. A broader neutralizing antibody against all the current VOCs and VOIs targets unique epitope of SARS-CoV-2 RBD. Cell Discov 2022;8:81. ArticlePubMedPMCPDF
- 14. Korber B, Fischer WM, Gnanakaran S, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020;182:812−7.ArticlePubMedPMC
- 15. Ahmad W, Ahmad S, Basha R. Analysis of the mutation dynamics of SARS-CoV-2 genome in the samples from Georgia State of the United States. Gene 2022;841:146774. ArticlePubMedPMC
- 16. GISAID. CoVsurver: mutation analysis of hCoV-19 [Internet]. GISAID; 2023 [cited 2023 Apr 27]. Available from: https://gisaid.org/database-features/covsurver-mutations-app/.
- 17. Cleemput S, Dumon W, Fonseca V, et al. Genome Detective Coronavirus Typing Tool for rapid identification and characterization of novel coronavirus genomes. Bioinformatics 2020;36:3552−5.ArticlePubMedPMCPDF
- 18. Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 2019;20:1160−6.ArticlePubMedPMCPDF
- 19. Martin DP, Murrell B, Golden M, et al. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 2015;1:vev003. ArticlePubMedPMCPDF
- 20. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014;30:1312−3.ArticlePubMedPMCPDF
- 21. Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 2021;49(W1):W293−6.ArticlePubMedPMCPDF
- 22. Yang Z, Rannala B. Bayesian phylogenetic inference using DNA sequences: a Markov Chain Monte Carlo Method. Mol Biol Evol 1997;14:717−24.ArticlePubMed
- 23. Suchard MA, Lemey P, Baele G, et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1. 10. Virus Evol 2018;4:vey016.
- 24. Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 1985;22:160−74.ArticlePubMedPDF
- 25. Rambaut A, Drummond AJ, Xie D, et al. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 2018;67:901−4.ArticlePubMedPMC
- 26. Singh J, Rahman SA, Ehtesham NZ, et al. SARS-CoV-2 variants of concern are emerging in India. Nat Med 2021;27:1131−3.ArticlePubMedPDF
- 27. Zhan Y, Yin H, Yin JY. B.1.617.2 (Delta) variant of SARS-CoV-2: features, transmission and potential strategies. Int J Biol Sci 2022;18:1844−51.ArticlePubMedPMC
- 28. Chen J, Wei GW. Omicron BA.2 (B.1.1.529.2): high potential for becoming the next dominant variant. J Phys Chem Lett 2022;13:3840−9.ArticlePubMedPMCPDF
- 29. Ministry of Health, Royal Government of Bhutan and World Health Organization. The people’s pandemic: how the Himalayan Kingdom of Bhutan staged a world-class response to COVID-19 [Internet]. Ministry of Health, Royal Government of Bhutan and World Health Organization; 2022 [cited 2023 Nov 10]. Available from: https://www.moh.gov.bt/wp-content/uploads/ict-files/2022/09/The-People%E2%80%99s-Pandemic-How-the-Himalayan-Kingdom-of-Bhutan-staged-a-world-class-response-to-COVID-19.pdf.
- 30. Zaman K, Shete AM, Mishra SK, et al. Omicron BA.2 lineage predominance in severe acute respiratory syndrome coronavirus 2 positive cases during the third wave in North India. Front Med (Lausanne) 2022;9:955930. ArticlePubMedPMC
- 31. Wei Y, Silke JR, Aris P, et al. Coronavirus genomes carry the signatures of their habitats. PLoS One 2020;15:e0244025.ArticlePubMedPMC
- 32. Rahman MM, Kader SB, Rizvi SM. Molecular characterization of SARS-CoV-2 from Bangladesh: implications in genetic diversity, possible origin of the virus, and functional significance of the mutations. Heliyon 2021;7:e07866.ArticlePubMedPMC
- 33. Yadav R, Chaudhary JK, Jain N, et al. Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells 2021;10:821. ArticlePubMedPMC
- 34. Solanki K, Rajpoot S, Kumar A, et al. Structural analysis of spike proteins from SARS-CoV-2 variants of concern highlighting their functional alterations. Future Virol 2022;17:723−32.ArticlePubMedPMC
- 35. Tian F, Tong B, Sun L, et al. N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. Elife 2021;10:e69091.ArticlePubMedPMCPDF
- 36. Ching WY, Adhikari P, Jawad B, et al. Effect of Delta and Omicron mutations on the RBD-SD1 domain of the spike protein in SARS-CoV-2 and the Omicron mutations on RBD-ACE2 interface complex. Int J Mol Sci 2022;23:10091. ArticlePubMedPMC
- 37. Wang R, Chen J, Gao K, et al. Vaccine-escape and fast-growing mutations in the United Kingdom, the United States, Singapore, Spain, India, and other COVID-19-devastated countries. Genomics 2021;113:2158−70.ArticlePubMedPMC
- 38. Abbasian MH, Mahmanzar M, Rahimian K, et al. Global landscape of SARS-CoV-2 mutations and conserved regions. J Transl Med 2023;21:152. ArticlePubMedPMCPDF
- 39. Johnson MA, Jaudzems K, Wuthrich K. NMR structure of the SARS-CoV nonstructural protein 7 in solution at pH 6.5. J Mol Biol 2010;402:619−28.ArticlePubMedPMC
- 40. Kirchdoerfer RN, Ward AB. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat Commun 2019;10:2342. ArticlePubMedPMCPDF
- 41. Ministry of Health, Royal Government of Bhutan, Thimphu, Bhutan. National situational update on COVID-19 [Internet]. Ministry of Health, Royal Government of Bhutan, Thimphu, Bhutan; 2023 [cited 2023 Jun 9]. Available from: https://www.moh.gov.bt/national-situational-update-on-covid-19-279/ .
- 42. Nie Q, Li X, Chen W, et al. Phylogenetic and phylodynamic analyses of SARS-CoV-2. Virus Res 2020;287:198098. ArticlePubMedPMC
- 43. Li X, Zai J, Wang X, et al. Potential of large “first generation” human-to-human transmission of 2019-nCoV. J Med Virol 2020;92:448−54.ArticlePubMedPMCPDF
- 44. Li X, Zai J, Zhao Q, et al. Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. J Med Virol 2020;92:602−11.ArticlePubMedPMCPDF
- 45. Li X, Wang W, Zhao X, et al. Transmission dynamics and evolutionary history of 2019-nCoV. J Med Virol 2020;92:501−11.ArticlePubMedPMCPDF
- 46. Dorji T, Tamang ST. Bhutan’s experience with COVID-19 vaccination in 2021. BMJ Glob Health 2021;6:e005977.ArticlePubMedPMC
- 47. Tsheten T, Tenzin P, Clements AC, et al. The COVID-19 vaccination campaign in Bhutan: strategy and enablers. Infect Dis Poverty 2022;11:6. ArticlePubMedPMCPDF
 Cite
Cite