Articles
- Page Path
- HOME > Osong Public Health Res Perspect > Volume 11(3); 2020 > Article
-
Original Article
Genome-Wide Identification and Characterization of Point Mutations in the SARS-CoV-2 Genome -
Jun-Sub Kima
 , Jun-Hyeong Janga, Jeong-Min Kima, Yoon-Seok Chunga, Cheon-Kwon Yoob, Myung-Guk Hana
, Jun-Hyeong Janga, Jeong-Min Kima, Yoon-Seok Chunga, Cheon-Kwon Yoob, Myung-Guk Hana -
Osong Public Health and Research Perspectives 2020;11(3):101-111.
DOI: https://doi.org/10.24171/j.phrp.2020.11.3.05
Published online: May 14, 2020
aDivision of Viral Diseases, Center for Laboratory Control of Infectious Diseases, Korea Centers for Disease Control and Prevention, Cheongju, Korea
bCenter for Laboratory Control of Infectious Diseases, Korea Centers for Disease Control and Prevention, Cheongju, Korea
- *Corresponding author: Myung-Guk Han Division of Viral Diseases, Center for Laboratory Control of Infectious Diseases, Korea Centers for Disease Control and Prevention, Cheongju, Korea E-mail: mghan@korea.kr
• Received: April 16, 2020 • Accepted: April 28, 2020
Copyright © 2020 Korea Centers for Disease Control & Prevention
This is an open access article under the CC BYNC-ND license (http://creativecommons.org/licenses/by-nc/4.0/).
Abstract
-
Objectives
- Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged in Wuhan, China, in December 2019 and has been rapidly spreading worldwide. Although the causal relationship among mutations and the features of SARS-CoV-2 such as rapid transmission, pathogenicity, and tropism, remains unclear, our results of genomic mutations in SARS-CoV-2 may help to interpret the interaction between genomic characterization in SARS-CoV-2 and infectivity with the host.
-
Methods
- A total of 4,254 genomic sequences of SARS-CoV-2 were collected from the Global Initiative on Sharing all Influenza Data (GISAID). Multiple sequence alignment for phylogenetic analysis and comparative genomic approach for mutation analysis were conducted using Molecular Evolutionary Genetics Analysis (MEGA), and an in-house program based on Perl language, respectively.
-
Results
- Phylogenetic analysis of SARS-CoV-2 strains indicated that there were 3 major clades including S, V, and G, and 2 subclades (G.1 and G.2). There were 767 types of synonymous and 1,352 types of non-synonymous mutation. ORF1a, ORF1b, S, and N genes were detected at high frequency, whereas ORF7b and E genes exhibited low frequency. In the receptor-binding domain (RBD) of the S gene, 11 non-synonymous mutations were observed in the region adjacent to the angiotensin converting enzyme 2 (ACE2) binding site.
-
Conclusion
- It has been reported that the rapid infectivity and transmission of SARS-CoV-2 associated with host receptor affinity are derived from several mutations in its genes. Without these genetic mutations to enhance evolutionary adaptation, species recognition, host receptor affinity, and pathogenicity, it would not survive. It is expected that our results could provide an important clue in understanding the genomic characteristics of SARS-CoV-2.
- Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged in Wuhan, China, in late December 2019. Since then, it has rapidly spread across the world, and was finally declared as a Public Health Emergency of International Concern (PHEIC) by the World Health Organization on January 30th, 2020 [1].
- SARS-CoV-2 is taxonomically classified under Nidovirales order, Coronaviridae family, Coronavirinae subfamily, and betacornoavirus genus. It is an enveloped virus with nonsegmented, positive-sense, single-stranded RNA. Although SARS-CoV-2 presents with a lower pathogenicity than severe acute respiratory syndrome coronavirus (SARS-CoV) which emerged in 2002–2003, and Middle-East respiratory syndrome coronavirus (MERS-CoV) which emerged in 2012, it reveals more rapidly human-to-human transmission [2].
- The genome of SARS-CoV-2 consists of non-segmented RNA that includes a 5’ untranslated region (UTR), structural proteins, non-structural proteins, several accessory proteins (open reading frames), and a 3’ UTR. The ORF1ab of several ORFs is proteolytically cleaved into 16 putative non-structural proteins (nsp1-16) for genome maintenance and replicase complex formation in viral replication. The structural proteins essential in viral particles include the spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins. The receptor-binding domain (RBD) of the S protein is crucial for binding directly to the human receptor ACE2, inducing viral entry, and determining host tropism and transmission capacity [3-5]. The S protein is cleaved into 2 subunits (S1 and S2). The S1 subunit directly recognizes and attaches to human receptor ACE2, while S2 fuses the host cell membrane with viral membranes allowing entry of SARS-CoV-2 [6]. In general, RNA viruses like SARS-CoV-2 undergo rapid mutation, enabling evolutionary and genetic diversity which result in alterations such as viral transmissibility, receptor affinity, host tropism, and pathogenicity.
- In recent years, several studies based on mutation analysis of SARS-CoV-2 genome have attempted to understand phylogenetic relationships, host infectivity, human-to-human transmission, viral tropism, and pathogenicity of SARS-CoV in humans. Firstly, the comparative evolutionary diversity in point mutations (synonymous-non-synonymous mutations) are suggestive that SARS-CoV-2 should to be classified into 3 major clades (S, G, and V) and other clades according to amino acid changes [7-9]. Secondly, the high affinity and stable structure of RBD/ACE2 have been associated with amino acid variations in the RBD such as the high affinity group (N354D, D364Y, V367F, and W436R) [10], and the high ACE2-binding affinity and stability group (484-NGVEGFN-490, Q496N, and Q496Y) [11]. Thirdly, the deletion of 382 nucleotides towards the 3’ end of the viral genome may have an impact on viral phenotype [12], and the QTQTN motif adjacent to the polybasic cleavage site (RRAR, chain of amino acids) at the bridge between S1 and S2 may be related to host adaptation [13]. In addition, insertion of the RRAR which has been well known to determine high or low pathogenicity in avian influenza virus may be important in determining transmissibility and pathogenesis of SARS-CoV-2 [14]. Finally, primer-template mismatch has been known to affect the stability and functionality of polymerase. In particular, the primer-template mismatch located in the primer 3’ end region can interfere with polymerase active sites, and this may have a significant impact on the accuracy of the molecular diagnosis using primers or probes [15].
- Therefore, we analyzed the mutations of the SARS-CoV-2 genome by focusing on phylogenetic evolution, RBD region, deletion mutations in polybasic cleavage site, and primer-template mismatches in the genome. Although the mechanisms responsible for rapid transmission, pathogenicity, and tropism in SARS-CoV-2 remain unclear, identification of mutations in the SARS-CoV-2 genome may help to interpret the high infectivity of the virus with the host.
Introduction
- The set of 4,254 SARS-CoV-2 genome sequences and acknowledgment files were downloaded from the EpiCoV browser (https://epicov.org/epi3) of the GISAID [16]. The raw data were processed by removing unnecessary genome sequences with low-quality reads, base calling errors, unsolved nucleotides as “N”, and small gaps. To investigate the genomewide phylogenetic analysis, we recombined 12 coding sequences (ORF1a, ORF1b, S, M, E, N, ORF3, ORF6, ORF7a, ORF7b, ORF8, and ORF10), excluding 5′ and 3′ UTR, low-quality sequences, and strains with high sequence similarity within the same clade. As a result of the processing, a reference genome (hCoV-19/Wuhan-Hu-1/2019, EPI_ISL_402125, 29,903 bp) and 178 fully complete genomes were collected. Phylogenetic analysis was performed to identify evolutionary relationships across the genome by using the MEGA [17] with parameters such as neighbor joining method, bootstrap 1,000 replications for the phylogeny test, Kimura 2-parameter as a substitution model, and pairwise deletion as gap/missing data treatment. For identifying the types of point mutation (synonymous and non-synonymous mutation) from 4,254 sequences, we separately extracted 12 fully coding sequences (CDSs) determining the sequence of amino acids in a protein from the genomes, and then conducted an in-house program based on Perl computer programming language to analyze mutations among the CDSs and the reference genome. In nomenclature for the replacement from one amino acid to a different amino acid in a gene, we illustrated it as the gene name surrounded by parenthesis after the substitution of amino acids. For example, D614G (S) means that an aspartic acid is converted into a glycine at amino acid position 614 of the S gene in comparison with the reference strain (hCoV-19/Wuhan-Hu-1/2019). For the comparative analysis of primer- and probe-template mismatches, we referred several lists published on the of Centers for Disease Control and Prevention (CDC) websites; https://www.who.int/docs/default-source/coronaviruse/uscdcrt-pcr-panel-primer-probes.pdf in CDC Atlanta, https://www.who.int/docs/default-source/coronaviruse/protocol-v2-1.pdf of the Charité Virology in Germany, https://www.who.int/docs/default-source/coronaviruse/peiris-protocol-16-1-20.pdf in school of public health at the university of Hong Kong, and https://www.who.int/docs/default-source/coronaviruse/real-time-rt-pcr-assays-for-the-detection-of-sars-cov-2-institut-pasteur-paris.pdf of the Institut Pasteur in Paris (Table 1).
Materials and Methods
- 1. Phylogenetic analysis of SARS-CoV-2 population
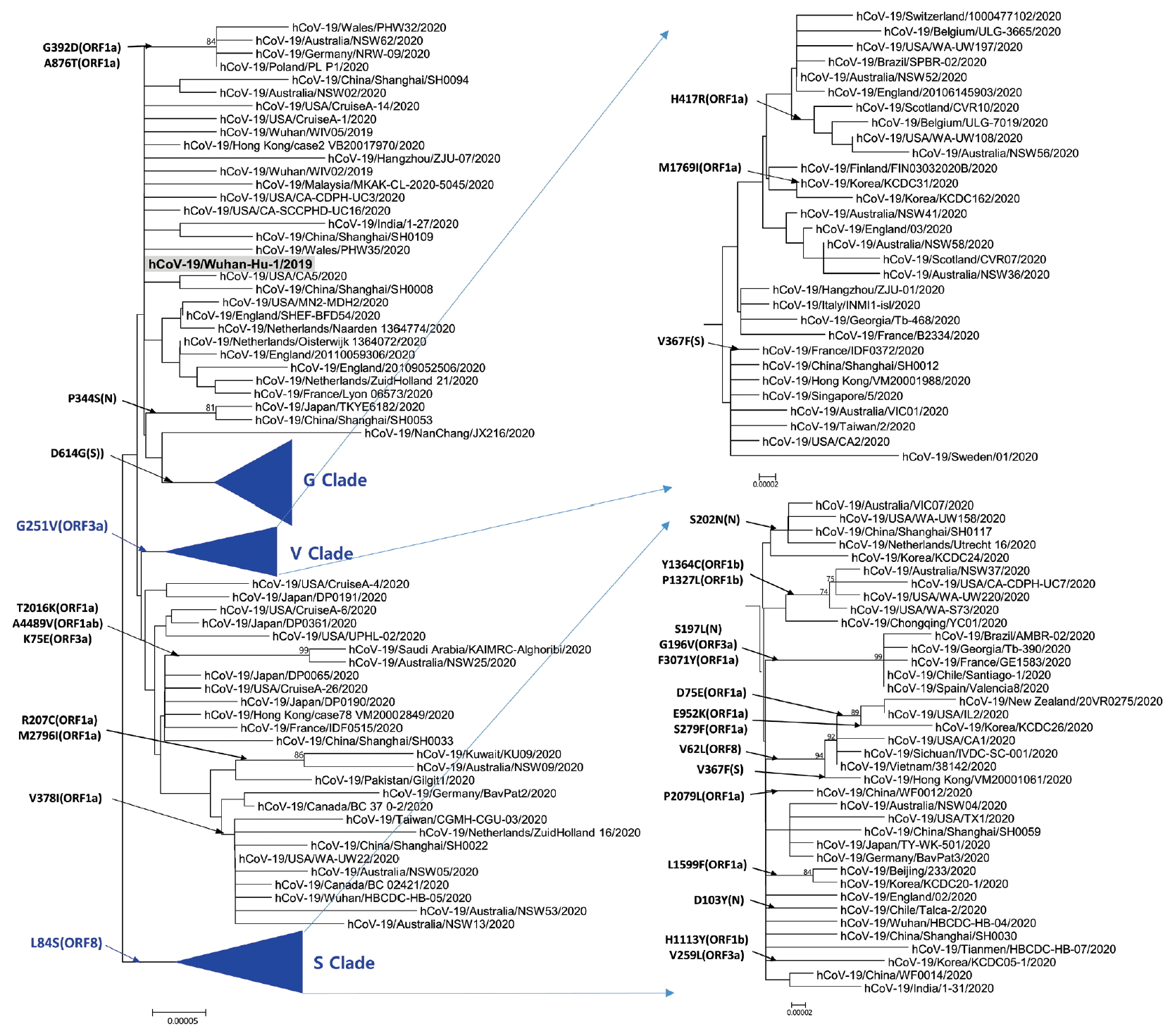
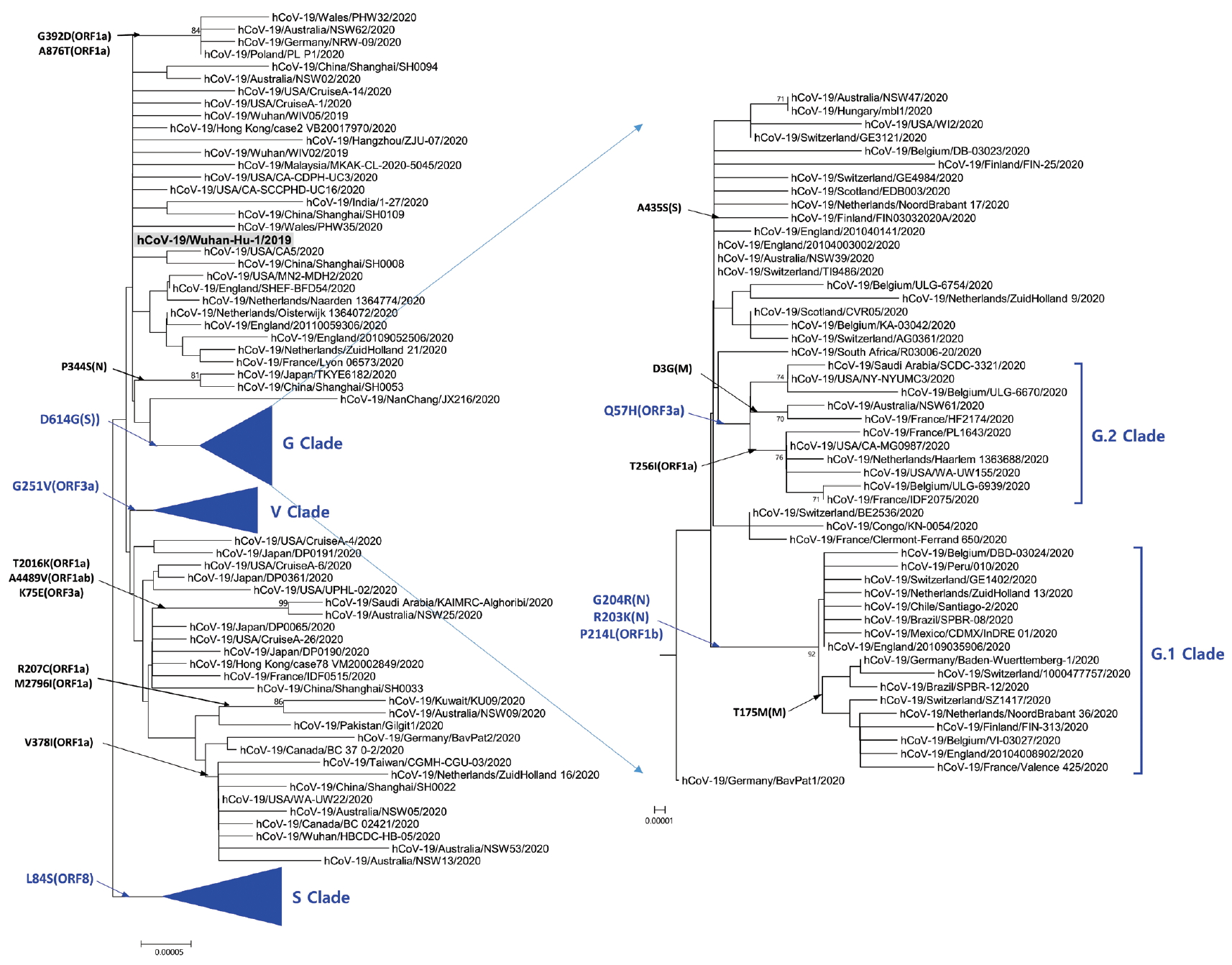
- Phylogenetic analysis was carried out with 178 representative strains to comprehend monophyletic distribution of the SARS-CoV-2 population. The phylogenetic tree showed 3 major clades (S, G, and V clades), similar to previous reports by GISAID. These clades (S, G, and V clades) were determined by the mutations [L84S (ORF8), G251V (ORF3a), and D614G (S)], respectively (Figure 1). Of the three mutations determining the clades, the significant D614G(S) in G clade is located in the adjacent polybasic cleavage site, but its actual function on RDB/ACE2 affinity is unclear. In addition to the three clades, two subclades belonging G clade were observed and they were named G.1, and G.2 clades referring to their parent clade name. The G.1 and G.2 subclades were determined by three mutations [G204R (N), R203K (N), and P214L (ORF1b)], and one mutation [Q57H (ORF3a)], respectively (Figure 2). In this respect, the derivation of G.1 and G.2 subclades from the G clade shows that a clade can be also determined by one or more mutations.
- 2. Frequency and types of mutation in SARS-CoV-2 genomes
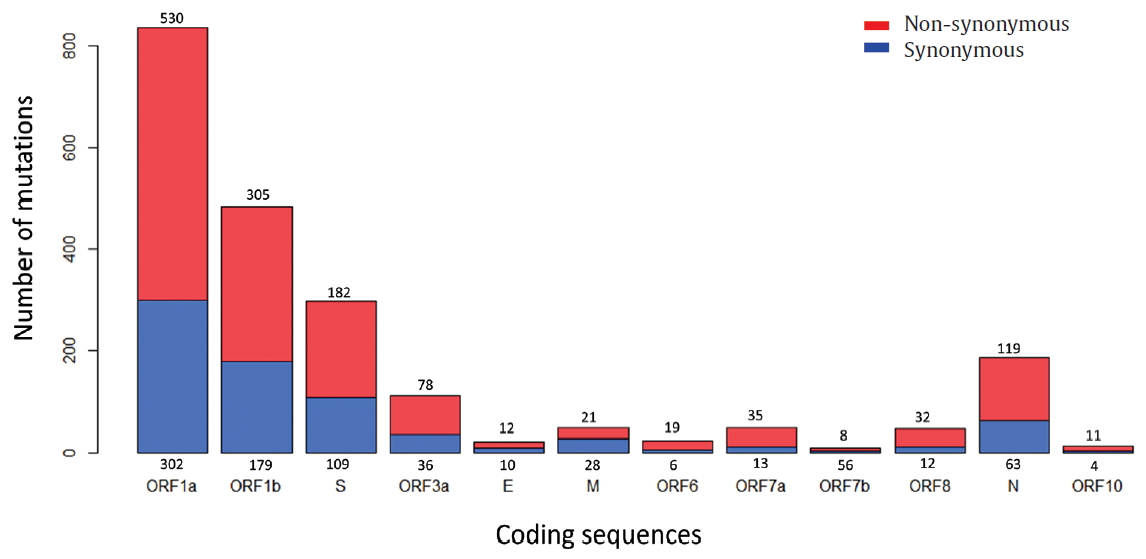
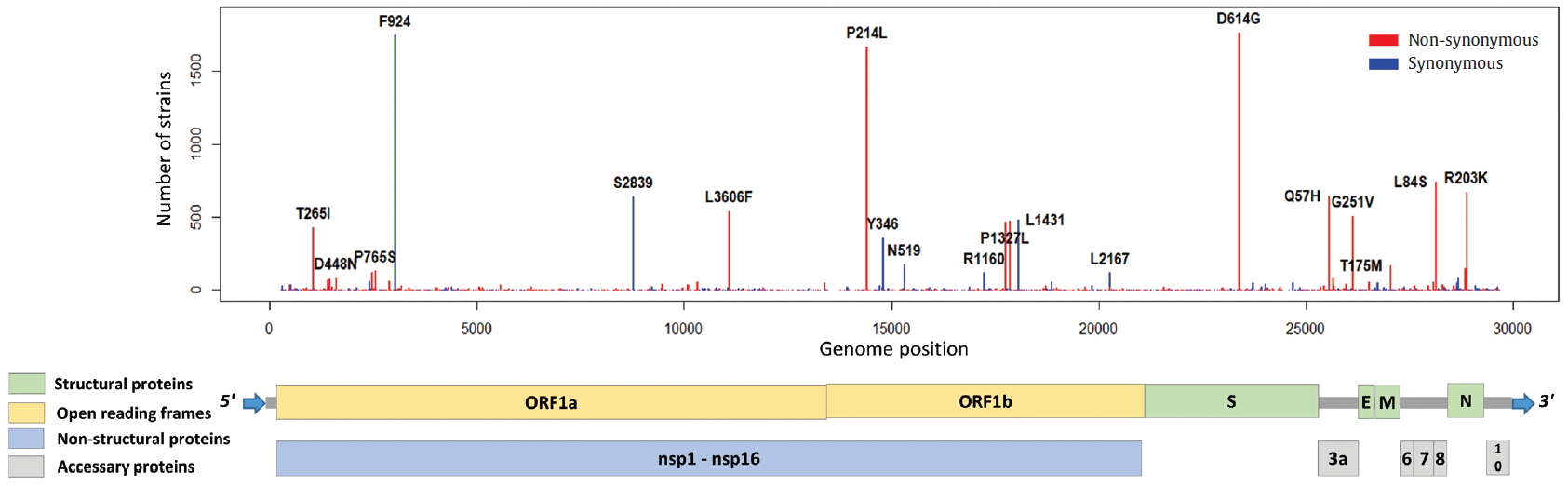
- To identify the number and the types of mutation across a total of SARS-CoV-2 strains, the 12 different types of CDS from the 4,254 strains were completely extracted in accordance with the genomic positions presented by the NCBI’s GenBank format file (sequence ID: NC_045512.2). The unsuitable sequences with base calling errors, unsolved nucleotides as “N”, and undefinable gaps were excluded. A total of 47,176 CDSs were gathered from ORF1a, ORF1b, S, M, E, N, ORF3, ORF6, ORF7a, ORF7b, ORF8, and ORF10. We identified 767 types of synonymous and 1,352 types of non-synonymous mutation from them. Genes with high frequency mutations were ORF1a, ORF1b, S, N, and ORF3a. ORF1a showed the highest frequency mutations containing 302 types of synonymous and 530 types of non-synonymous mutation (Table 2, Figure 3). The non-synonymous mutations with high frequency in the genes were representative mutations in the clades: L84S (ORF8) in S clade, D614G (S) in G clade, G251V (ORF3a) in V clade, P214L (ORF1b), R203K (N) and G204R (N) in G.1 clade, and Q57H (ORF3a) in G.2 clade (Figures 4 and 5).
- 3. Mutations in ORF1a, ORF1b, and RdRp
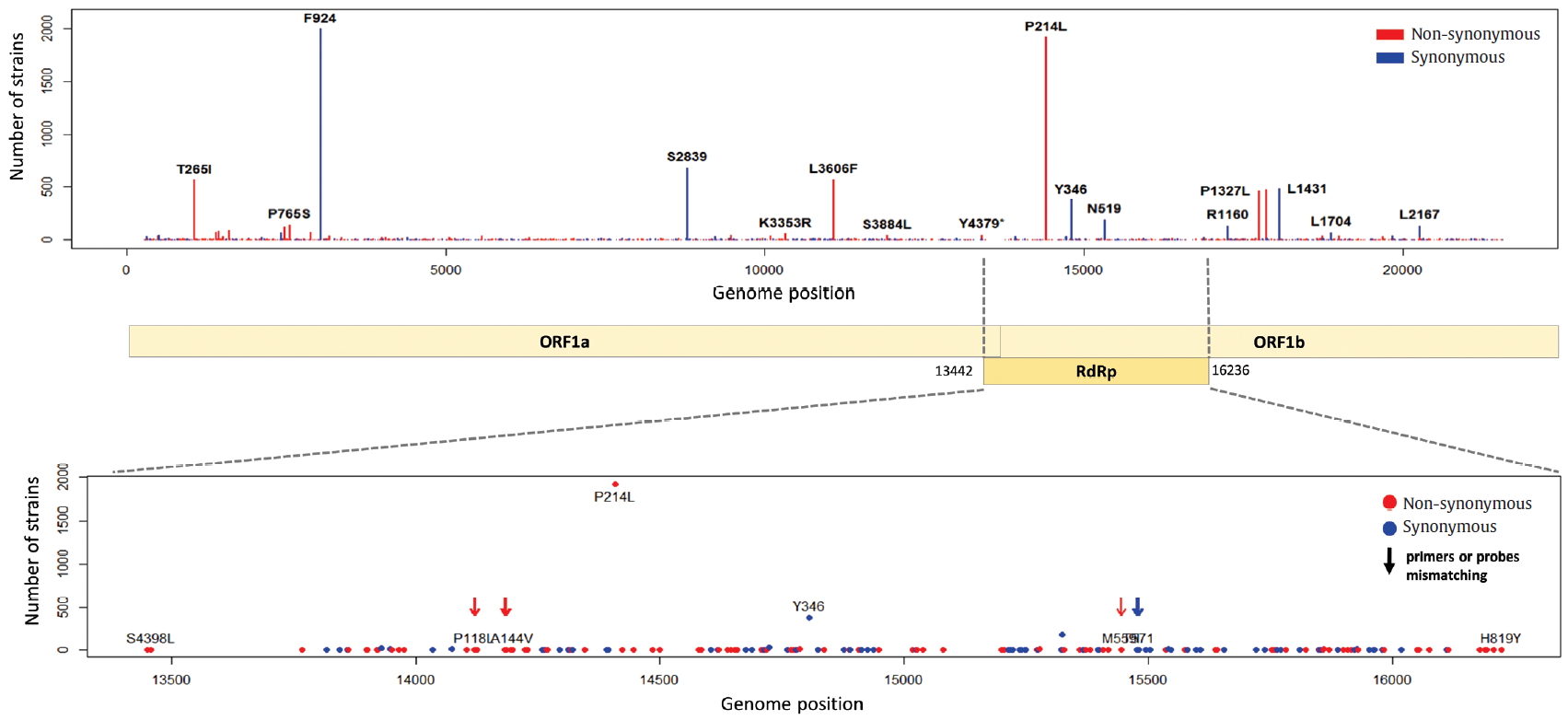
- ORF1a and ORF1b encode replicase polyproteins essential component of the viral RNA replication. Therefore, conserved structure of them have been the focus of antiviral drugs [18]. RNA-dependent RNA polymerase (RdRp) located between ORF1a and ORF1b is an important component for application and translation and has a functionally conserved region known as high sequence similarity [19]. The conserved region of RdRp has been widely used as a target for designing primers and probes based on RT-PCR technology in genetic diagnosis of coronavirus disease 2019 (COVID-19). In this regard, the mutations from ORF1a and ORF1b were analyzed, and we identified 302 types of synonymous and 530 types of non-synonymous mutation in ORF1a, and 179 types of synonymous and 305 types of non-synonymous mutation in ORF1b. Also, a total of 144 types of mutation were identified in the RdRp region corresponding to the 13,442 to 16,236 genomic position, of which 56 types of synonymous and 88 types of non-synonymous mutation were detected (Figure 6). Since the non-synonymous mutation with the highest frequency of in the RdRp region was P214L which determines the G.1 subclade together with G204R and R203K of the N gene, it was necessary to further study the functional interaction amongst these mutations of the ORF1a and N gene. On one hand, 9 types of primer-template mismatch were identified in the RdRp region. Although it is not understood how these mismatches affect the stability of primer-template complexes, it is necessary to determine the interrelationships.
- 4. Mutations in S gene, receptor-binding domain and polybasic cleavage site
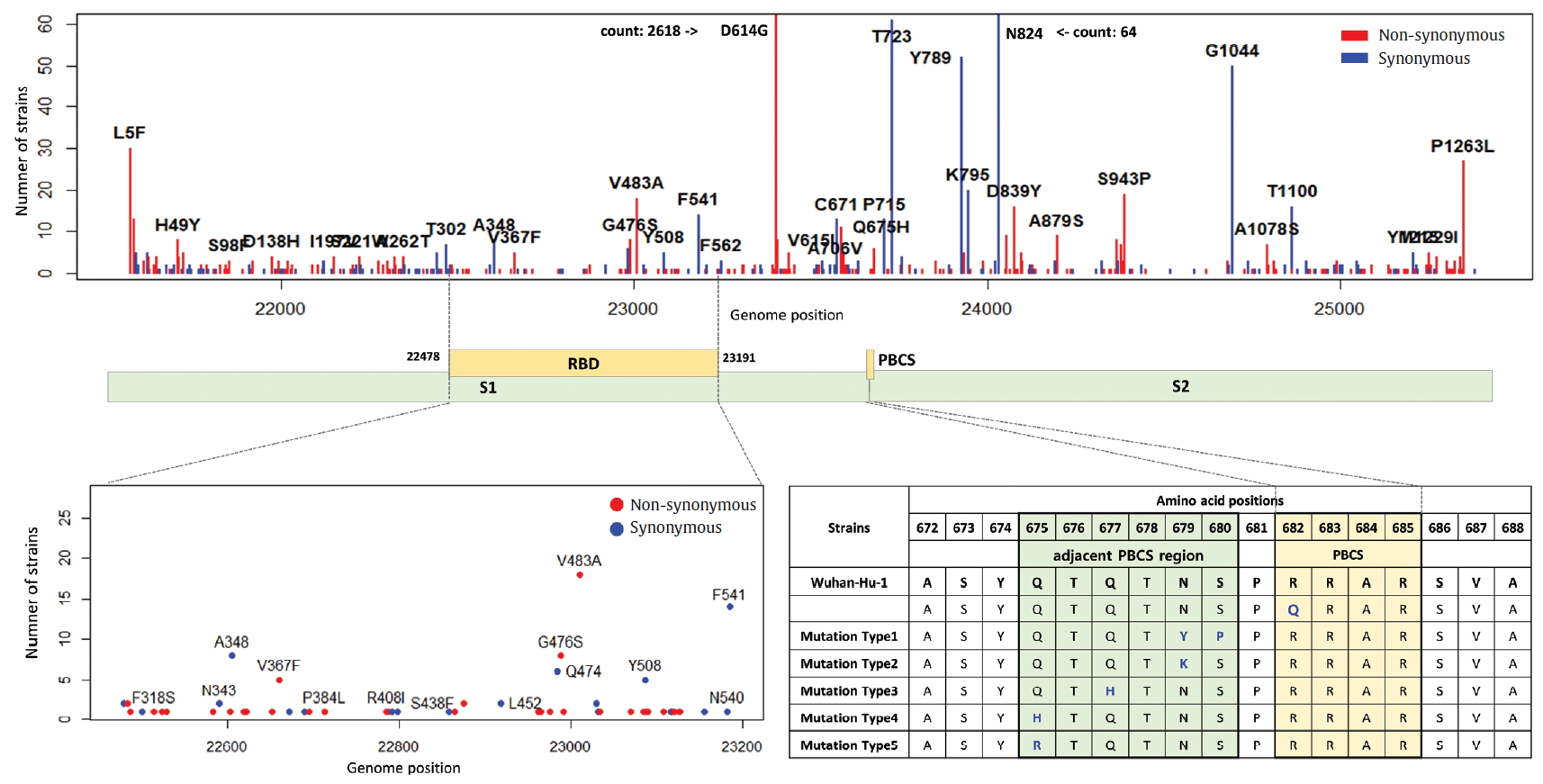
- The S gene of SARS-CoV-2 plays an important role in escaping the immune system of host. The RBD in the S gene is directly or indirectly concerned with the binding and affinity to ACE2. The strong binding between RBD and ACE2 may have led to the worldwide transmission and rapid infectivity of SARS-CoV-2. On the other hand, the RRAR, which is located between S1 and S2 (681-685 amino acid position of the S gene), may be related to the pathogenicity of SARS-CoV-2. Therefore, it is might be inferred from the mutations in the RBD and the polybasic cleavage site in the S gene that the mutations will provide important clues to understanding immunity and pathogenicity of SARS-CoV-2. We identified 109 types of synonymous and 182 types of non-synonymous mutation in the S gene (Figure 7). D614G is a major non-synonymous mutation in G clade and accounts for 1,764 (13.0%) in a total of 13,537 non-synonymous mutations. Above all, it is located in the S1-S2 junction region near the polybasic cleavage site, but its biochemical and structural relationships with ACE2 has been unclear so far. In the RBD region (22,478-23,191 genomic position), we found 12 types of synonymous and 27 types of non-synonymous mutation. Among them, the D467V, I468F, I468T, I472V, G476S, S477G, V483A, P491R, Y508H, R509K, and V510L were located within the zone (443-510 amino acid position of the S gene) adjacent to ACE2. In particular, the V483A and G476S mutations have previously been reported to be related to human receptor-binding affinity in MERS and SARS-CoV research [20, 21]. We detected a R682Q mutation from the RRAR, where arginine residue was replaced by glutamine residue. The arginine residue is not only electrically charged but also strongly basic, while the glutamine residue is polar uncharged. Therefore, it is necessary to study how this biochemical difference may affect functional and structural changes of the proteins of S1 or S2. According to a recent study, the fact that a deletion of the QTQTN amino acid motif near the polybasic cleavage site is related to adaptation of SARS-CoV-2 has been reported. Hence, we tried to confirm whether some deletions occurred in this region (675-679 amino acid position of the S gene). We identified no deletion of QTQTN in this region, but confirmed 5 types of non-synonymous mutation (Type 1 with N679Y and S680P, Type 2 with N679K, Type 3 with Q677H, Type 4 with Q675H, and Type 5 with Q675R). Except for Type 1, Type 2 to 5 had a common feature where glutamine and asparagine with a polar uncharged side chain were converted into lysine, histidine, or arginine with basic property. Although deletion of the QTQTN motif was not identified near the polybasic cleavage site, it may be informative to study the functional significance of the five mutations in this region.
- 5. Mutations in E, M, N and open reading frames encoding accessory proteins
- The E, M, ORF6, ORF7a, ORF7b, ORF8, and ORF10 are less frequent mutations than other genes. ORF3a is a novel short positive protein essential to viral adaptation in vitro and is involved in the viral pathogenicity. The major types of synonymous mutation of ORF3a are Q57H and G251V, which are significant mutations in determining G.2 clade, and V clade, respectively. It is inferred from this result that Q57H and G251V may have a positive effect on viral adaptation. In the E gene known to be involved in viral budding along with the M gene, we confirmed a relatively small number of mutations in it. This means that there are high conserved regions, so it seems reasonable to infer that the preserved sequence is important to encode protein participating in viral reproduction. Like the region of RdRp, the E gene has been also used to design primers and probes for the diagnosis of SARS-CoV-2. We identified 10 types of synonymous and 12 types of non-synonymous mutation in the E gene (Figure 8). Of these mutations, 7 primer-template mismatch mutations (3 types of synonymous and 4 types of non-synonymous mutation) occurred. The M gene which is associated with cellular immunogenicity, showed 28 types of synonymous and 21 types of non-synonymous mutation, of which mutations with high frequency were observed as T175M and D3G. Several types of synonymous mutation in ORF6, ORF7, and ORF10 were identified, but mutations with high frequency were not observed. However, L84S in ORF8 was identified as a mutation with high frequency. The N gene packaging of the viral RNA genome into the medical ribonucleocapsid has been regarded as a candidate target region for primers and probes like the E and RdRp genes. In the N gene, we detected 63 types of synonymous and 119 types of non-synonymous mutation. Among them, R203K and G204R showed high frequencies in G.2 clade, together with P214L (ORF1b). In addition, 51 primer-template mismatch mutations (17 types of synonymous and 34 types of non-synonymous mutation) occurred.
Results
- From the non-synonymous mutation is overall more frequent than the synonymous mutations in the set of 4,254 SARS-CoV-2 genome sequences, it is inferred from this results that the evolution of the SARS-CoV-2 has been accepted to be positive selection. In consequence, several point mutations may directly or indirectly influence the interaction between SARS-CoV-2 and human, and the diversity of mutations in SARS-CoV-2 may enhance the evolution of the virus towards rapid transmission. Based on our analysis, the D614G (S) mutation of G clade, which revealed the highest frequent in our phylogenetic analysis, may have a positive advantage in natural selection. According to the frequency of mutations within SARS-CoV-2 strains, ORF1a, ORF1b, S, and N genes were shown at high frequency, and they may advantageously evolve to adapt to not only external interactions with host cells (such as recognizing a cell surface receptor, attaching to the host receptor, and fusing with cellular membranes), but also internal interactions in host cells (such as replicating and transcribing viral genome, and budding by cellular exocytosis). Although D614G in the S gene is not a mutation within RBD, the fact that it occurred as the highest frequency in all SARS-CoV-2 genomes suggests it may be related to host infection and transmission. Therefore, we believe that it is necessary to continuously monitor the accumulation of mutations and to further study how these mutations affect receptor affinity, propagation ability, and pathogenicity. On the other hand, RdRp, E, and N genes are the target genes for designing primers and probes in RT-PCR-based SARS-CoV-2 diagnosis, owing to their high sequence conservation. It has not been known how the primer-template mismatches affect the accuracy and precision of the genetic diagnosis of COVID-19, but we suggest that it is desirable to avoid the variable hotspot regions as much as possible. Our results about genomic mutations of SARS-CoV-2 strains may be helpful for interpreting the potential relationships of pathogenicity, infectivity, and transmission between SARS-CoV-2 and human host.
Discussion
-
Acknowledgements
- We acknowledge the incessant devotion and actively hard work of the staffs in the Division of Viral Diseases Research from the Korea Centers for Disease Control and Prevention. This study was funded by Korea Centers for Disease Control and Prevention (no.: 4800-4837-301).
Figure. 1.Phylogenetic tree of SARS-CoV-2. This tree was performed by using the MEGA with parameters such as neighbor joining method, bootstrap 1,000 replications for the phylogeny test, Kimura 2-parameter for substitution model, and pairwise deletion for gap/missing data treatment. S and V clades determined by L84G (ORF8) and G251V (ORF3a), respectively. The reference strain (hCoV-19/Wuhan-Hu-1/2019) is indicated by bold letters on a light gray background. The notation of amino acid substitutions used here means replacements from amino acid of the reference strain on left to a difference amino acid of the corresponding strain on right. MEGA = molecular evolutionary genetics analysis.
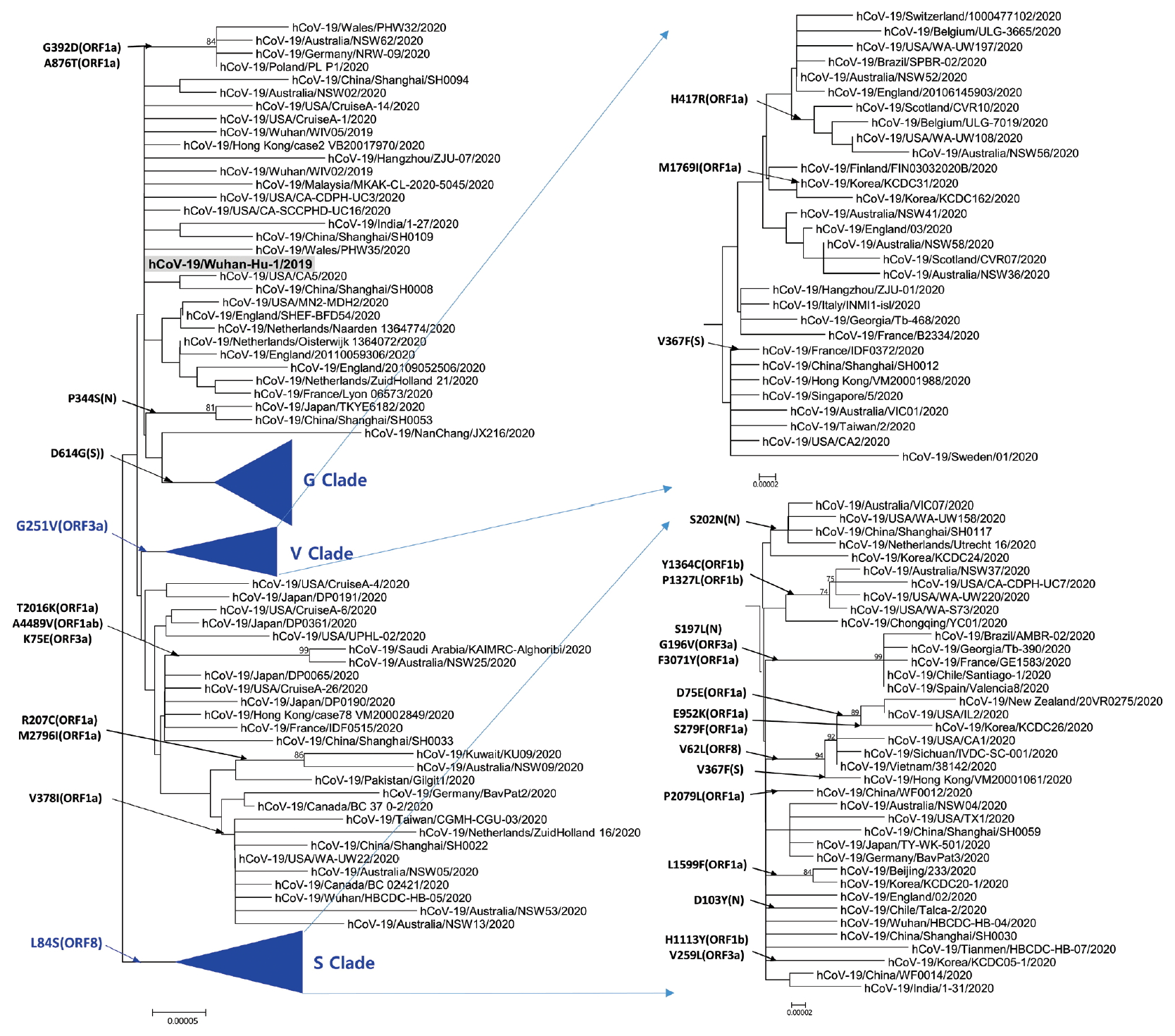
Figure. 2.Phylogenetic tree of G clade and its subclades. G Clade determined by D614G (S) was classified into G.1 clade, G.2 clade, and other strains. G.1 and G.2 clades share Q57H (ORF3a) and G204R (N),
R203K (N), together with P214L (ORF1b), respectively. The reference strain (hCoV-19/Wuhan-Hu-1/2019) is indicated by bold letters on a light gray background. The notation of amino acid substitutions used here means replacements from amino acid of the reference strain on left to a difference amino acid of the corresponding strain on right.

Figure. 3.Types of mutation distribution in 12 coding sequences. This figure summarizes the distribution of point mutations on 12 coding sequences (ORF1a, ORF1b, S, ORF3a, E, M, ORF6, ORF7a, ORF7b, ORF8, N, and ORF10). Bars filled with red color indicates non-synonymous mutations and bars filled with blue color indicates synonymous mutation. Number above and below bars show the type of mutation.

Figure. 4.Frequency of mutations in coding sequences. (A) indicates frequency of 5,940 synonymous mutations (767 types of synonymous mutation) in 47,176 coding sequences from 4,254 strains. (B) indicates frequency of 13,537 non-synonymous mutations (1,352 types of non-synonymous mutation) in them.

Figure. 5.Distribution of mutations in SARS-CoV-2 whole genome. The length of SARS-CoV-2 genome is 29,903 bases. SARS-CoV-2 genome is composed of 5’ UTR, ORF1a, ORF1b, S, E, M, N, accessary proteins (ORF3a, ORF6, ORF7, ORF8, and ORF10), and 3’ UTR. The ORF1a and ORF1b encodes from nsp1 to nsp16 proteins.
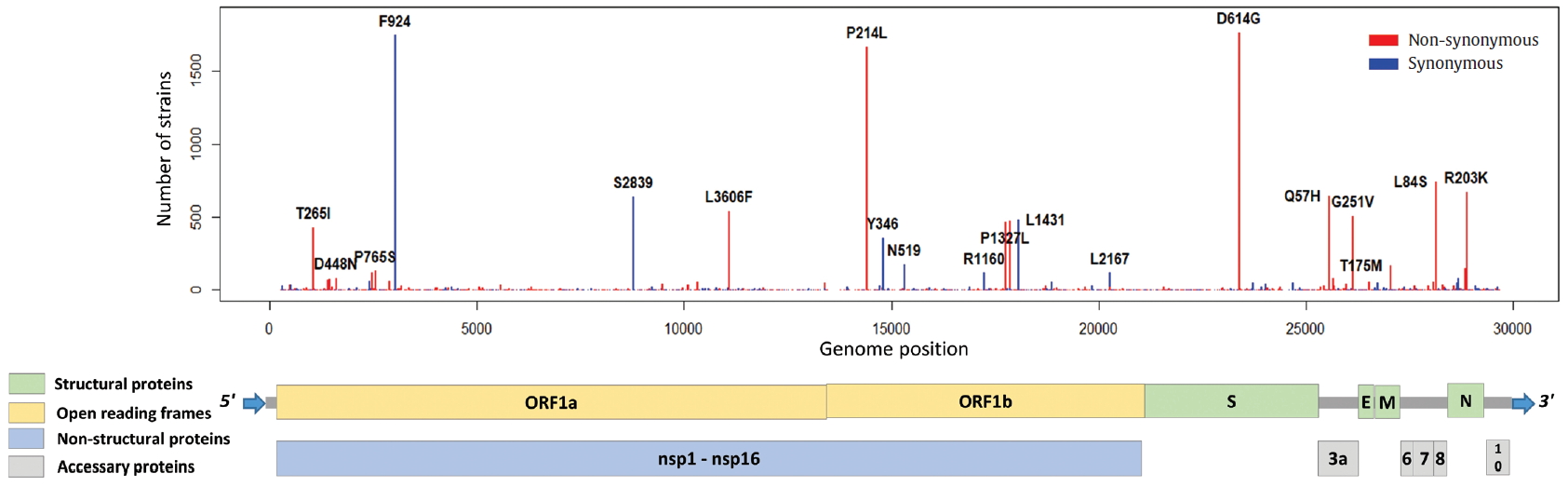
Figure. 6.Distribution of mutations and primer-template mismatches in ORF1a and ORF1b. The genomic positions (13,442-16,236) of RNA-dependent RNA polymerase (RdRp) was based on NCBI ID NC_045512.2. The primer- and probe-template regions were derived from the lists published on the Centers for Disease Control and Prevention (CDC).
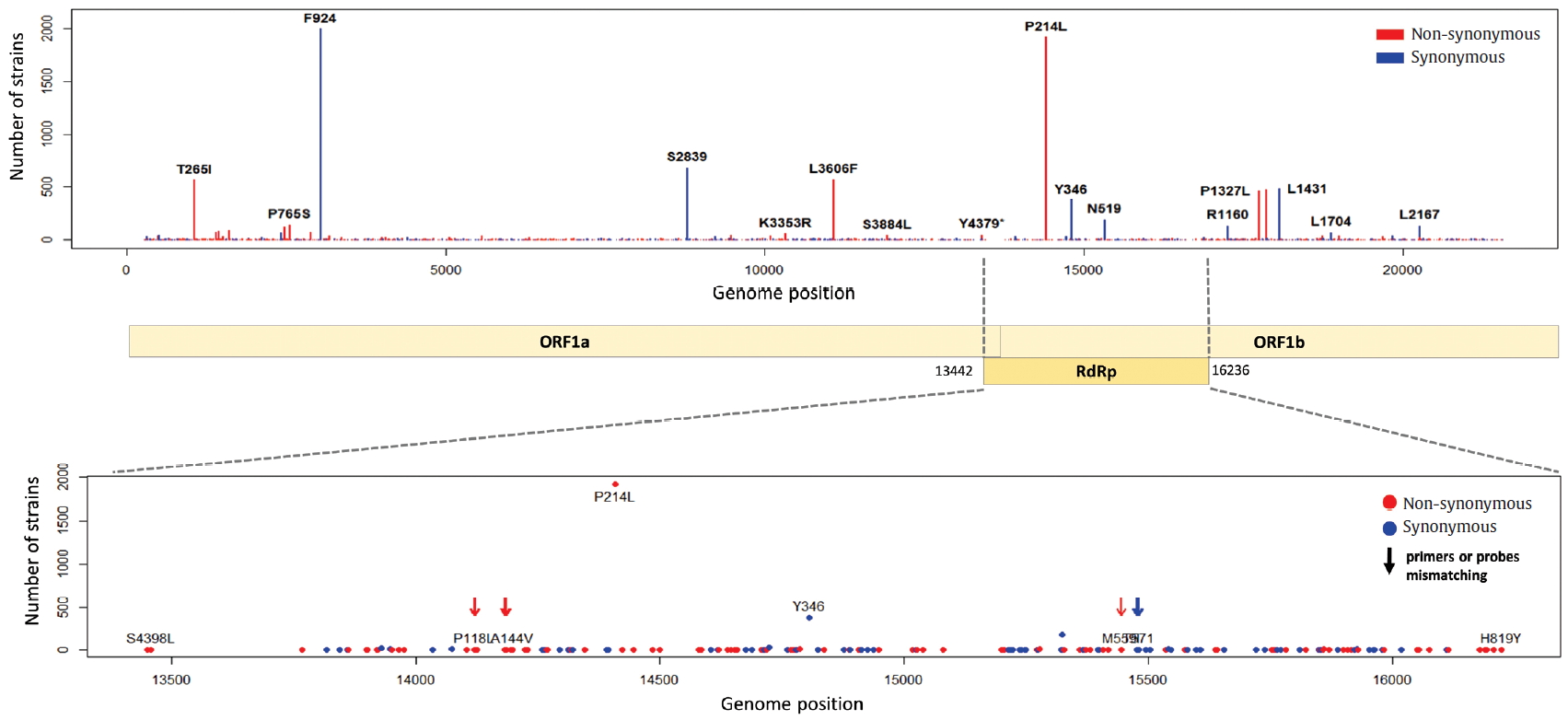
Figure. 7.Distribution of mutations in receptor-binding domain and polybasic cleavage site within S gene. The genomic positions (22,478-23,191) of receptor-binding domain (RBD) and the amino acid position (682-685) of polybasic cleavage site (PBCS) were based on the reference [4], and the reference [14], respectivity.
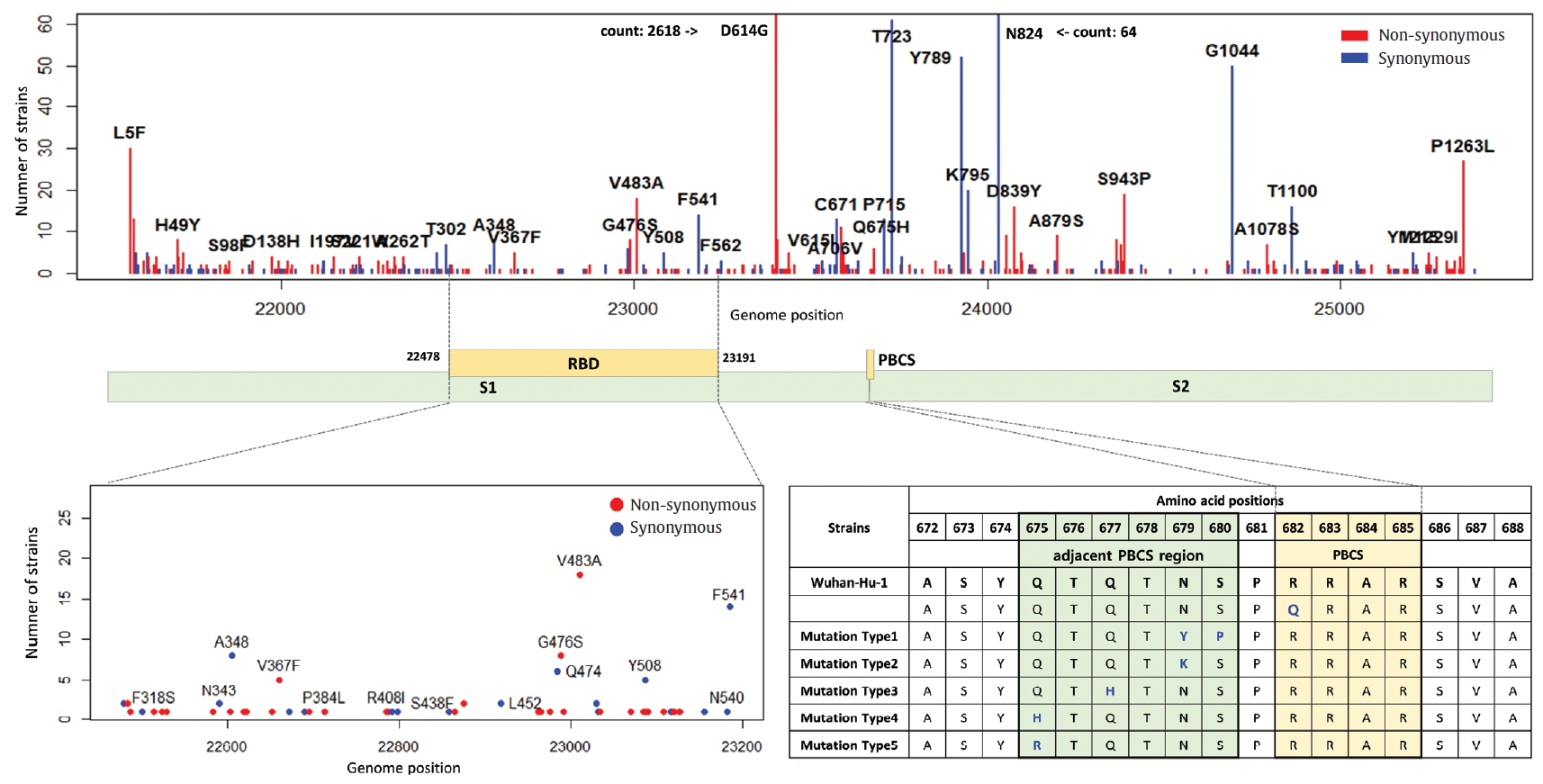
Figure. 8.Distribution of mutations and primer-template mismatches in ORF3a, E, M, ORF6, ORF7a, ORF7b, ORF8, N, and ORF10. The genomic positions (13,442-16,236) of RNA-dependent RNA polymerase (RdRp) was based on NCBI ID NC_045512.2. The primer- and probe-template regions were retrieved from the lists published on the Centers for Disease Control and Prevention (CDC).
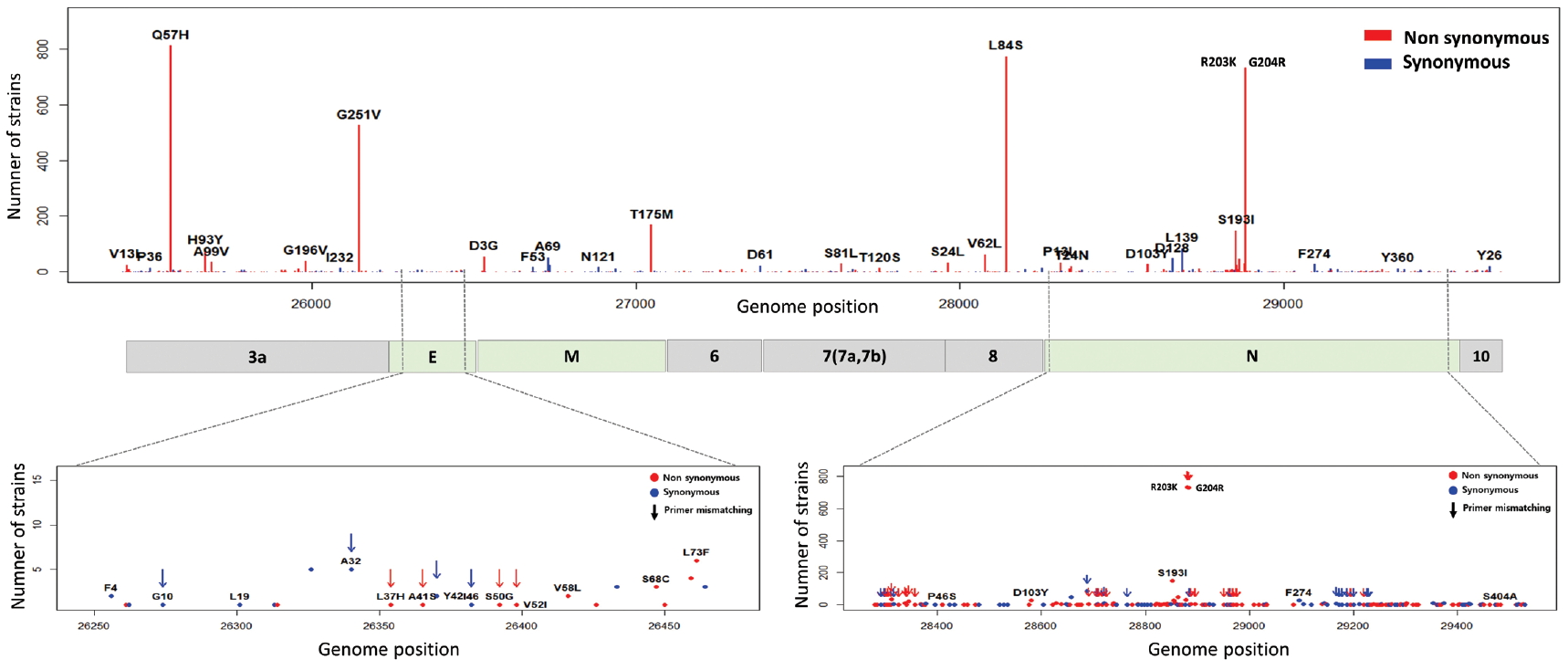
Table 1.Primers and probes for the detection of SARS-CoV-2 of global research institutions.
Table 2.Frequency of point mutations by 12 coding sequences.
|
CDS |
Genomic position* |
Point mutations |
||||
|---|---|---|---|---|---|---|
| Name | No. of strains |
No. of mutation |
Type of mutation |
|||
| Synonymous | Non-synonymous | Synonymous | Non-synonymous | |||
| ORF1a | 3,325 | 266-13,483 | 3,199 | 2,850 | 302 | 530 |
| ORF1b | 3,360 | 13,768-21,555 | 1,700 | 3,194 | 179 | 305 |
| S | 3,498 | 21,563-25,384 | 367 | 2,212 | 109 | 182 |
| ORF3a | 4,116 | 25,393-26,220 | 72 | 1,450 | 36 | 78 |
| E | 4,216 | 26,245-26,472 | 23 | 21 | 10 | 12 |
| M | 4,101 | 26,523-27,191 | 147 | 251 | 28 | 21 |
| ORF6 | 4,199 | 27,202-27,387 | 29 | 34 | 6 | 19 |
| ORF7a | 3,965 | 27,394-27,759 | 35 | 71 | 13 | 35 |
| ORF7b | 3,988 | 27,756-27,887 | 8 | 9 | 5 | 8 |
| ORF8 | 4,198 | 27,894-28,259 | 26 | 874 | 12 | 32 |
| N | 4,080 | 28,274-29,533 | 305 | 2,549 | 63 | 119 |
| ORF10 | 4,130 | 29,558-29,674 | 29 | 22 | 4 | 11 |
- 1. Wrapp D, Wang N, Corbett KS, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020;367(6483). 1260−3.ArticlePubMedPMC
- 2. Rehman SU, Shafique L, Ihsan A, et al. Evolutionary trajectory for the emergence of novel coronavirus SARS-CoV-2. Pathogens 2020;9(3). E240.Article
- 3. The Johns Hopkins Center for Health Security: nCoV Genetics [Internet]. [cited 2020 Feb 3]. Available from: http://www.centerforhealthsecurity.org/resources/COVID-19/COVID-19-fact-sheets/200128-nCoV-whitepaper.pdf.
- 4. Chan JF, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect 2020;9(1). 221−36.ArticlePubMedPMC
- 5. Ou X, Guan H, Qin B, et al. Crystal structure of the receptor-binding domain of the spike glycoprotein of human betacoronavirus HKU1. Nat Commun 2017;8:15216. ArticlePubMedPMCPDF
- 6. Li F, Li W, Farzan M, et al. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005;309(5742). 1864−8.ArticlePubMed
- 7. Tang X, Wu C, Li X, et al. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev 2020;[Epub ahead of print]. Epub 2020 Mar 3.ArticlePDF
- 8. Ceraolo C, Giorgi FM. Genomic variance of the 2019-nCoV coronavirus. J Med Virol 2020;92(5). 522−8.ArticlePubMedPMC
- 9. Zhang L, Yang JR, Zhang Z, et al. [Preprint] Genomic variations of SARS-CoV-2 suggest multiple outbreak sources of transmission. medRxiv: 2020.02.25.20027953. 2020. Available from: https://www.medrxiv.org/content/10.1101/2020.02.25.20027953v2.Article
- 10. Ou J, Zhou Z, Zhang J, et al. [Preprint] RBD mutations from circulating SARS-CoV-2 strains enhance the structure stability and infectivity of the spike protein. bioRxiv: 2020.03.15.991844. 2020. Available from: https://www.biorxiv.org/content/10.1101/2020.03.15.991844v1.
- 11. Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020;581(7807). 221−4.ArticlePDF
- 12. Su YCF, Anderson DE, Young BE, et al. [Preprint] Discovery of a 382-nt deletion during the early evolution of SARS-CoV-2. bioRxiv: 2020.03.11.987222. 2020. Available from: https://www.biorxiv.org/content/10.1101/2020.03.11.987222v1.Article
- 13. Liu Z, Zheng H, Yuan R, et al. [Preprint] Identification of a common deletion in the spike protein of SARS-CoV-2. bioRxiv: 2020.03.31.015941. 2020. Available from: https://www.biorxiv.org/content/10.1101/2020.03.31.015941v1.Article
- 14. Andersen KG, Rambaut A, Lipkin WI, et al. The proximal origin of SARS-CoV-2. Nat Med 2020;26(4). 450−2.ArticlePubMedPMCPDF
- 15. Stadhouders R, Pas SD, Anber J, et al. Effect of Primer-Template Mismatches on the Detection and Quantification of Nucleic Acids Using the 5′ Nuclease Assay. J Mol Diagn 2010;12(1). 109−17.ArticlePubMedPMC
- 16. Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill 2017;22(13). 30494. ArticlePubMedPMC
- 17. Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 2016;33(7). 1870−4.ArticlePubMedPMCPDF
- 18. Yin C. Genotyping coronavirus SARS-CoV-2: Methods and implications. Genomics 2020;[Epub ahead of print]. Epub 2020 Apr 27.Article
- 19. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020;579(7798). 265−9.ArticlePDF
- 20. Kleine-Weber H, Elzayat MT, Wang L, et al. Mutations in the Spike Protein of Middle East Respiratory Syndrome Coronavirus Transmitted in Korea Increase Resistance to Antibody-Mediated Neutralization. J Virol 2019;93(2). e01381−18.PubMedPMC
- 21. Wu K1, Peng G, Wilken M, et al. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J Biol Chem 2012;287(12). 8904−11.ArticlePubMedPMC
References
Figure & Data
References
Citations
Citations to this article as recorded by 
- Three-Dimensional Structural Stability and Local Electrostatic Potential at Point Mutations in Spike Protein of SARS-CoV-2 Coronavirus
Svetlana H. Hristova, Alexandar M. Zhivkov
International Journal of Molecular Sciences.2024; 25(4): 2174. CrossRef - Geographical distribution of host's specific SARS-CoV-2 mutations in the early phase of the COVID-19 pandemic
Mohammad Khalid, David Murphy, Maryam Shoai, Jonahunnatha Nesson George-William, Yousef Al-ebini
Gene.2023; 851: 147020. CrossRef - SARS-CoV-2 mutations on diagnostic gene targets in the second wave in Zimbabwe: A retrospective genomic analysis
C Nyagupe, L de Oliveira Martins, H Gumbo, T Mashe, T Takawira, KK Maeka, A Juru, LK Chikanda, AR Tauya, AJ Page, RA Kingsley, R Simbi, J Chirenda, J Manasa, V Ruhanya, RT Mavenyengwa
South African Medical Journal.2023; 113(3): 141. CrossRef - A low dose of RBD and TLR7/8 agonist displayed on influenza virosome particles protects rhesus macaque against SARS-CoV-2 challenge
Gerrit Koopman, Mario Amacker, Toon Stegmann, Ernst J. Verschoor, Babs E. Verstrepen, Farien Bhoelan, Denzel Bemelman, Kinga P. Böszörményi, Zahra Fagrouch, Gwendoline Kiemenyi-Kayere, Daniella Mortier, Dagmar E. Verel, Henk Niphuis, Roja Fidel Acar, Ivan
Scientific Reports.2023;[Epub] CrossRef - Detection of IgA and IgG Antibodies against the Structural Proteins of SARS-CoV-2 in Breast Milk and Serum Samples Derived from Breastfeeding Mothers
Karen Cortés-Sarabia, Vianey Guzman-Silva, Karla Montserrat Martinez-Pacheco, Jesús Alberto Meza-Hernández, Víctor Manuel Luna-Pineda, Marco Antonio Leyva-Vázquez, Amalia Vences-Velázquez, Fredy Omar Beltrán-Anaya, Oscar Del Moral-Hernández, Berenice Illa
Viruses.2023; 15(4): 966. CrossRef - Evaluation of antiviral drugs against newly emerged SARS-CoV-2 Omicron subvariants
Junhyung Cho, Younmin Shin, Jeong-Sun Yang, Jun Won Kim, Kyung-Chang Kim, Joo-Yeon Lee
Antiviral Research.2023; 214: 105609. CrossRef - Genetic Analysis and Epitope Prediction of SARS-CoV-2 Genome in Bahia, Brazil: An In Silico Analysis of First and Second Wave Genomics Diversity
Gabriela Andrade, Guilherme Matias, Lara Chrisóstomo, João da Costa-Neto, Juan Sampaio, Arthur Silva, Isaac Cansanção
COVID.2023; 3(5): 655. CrossRef - Natural selection shapes the evolution of SARS-CoV-2 Omicron in Bangladesh
Mohammad Tanbir Habib, Saikt Rahman, Mokibul Hassan Afrad, Arif Mahmud Howlader, Manjur Hossain Khan, Farhana Khanam, Ahmed Nawsher Alam, Emran Kabir Chowdhury, Ziaur Rahman, Mustafizur Rahman, Tahmina Shirin, Firdausi Qadri
Frontiers in Genetics.2023;[Epub] CrossRef - Unraveling the impact of ORF3a Q57H mutation on SARS-CoV-2: insights from molecular dynamics
Md. Jahirul Islam, Md. Siddik Alom, Md. Shahadat Hossain, Md Ackas Ali, Shaila Akter, Shafiqul Islam, M. Obayed Ullah, Mohammad A. Halim
Journal of Biomolecular Structure and Dynamics.2023; : 1. CrossRef - Genomic characterization of SARS-CoV-2 from Uganda using MinION nanopore sequencing
Praiscillia Kia, Eric Katagirya, Fredrick Elishama Kakembo, Doreen Ato Adera, Moses Luutu Nsubuga, Fahim Yiga, Sharley Melissa Aloyo, Brendah Ronah Aujat, Denis Foe Anguyo, Fred Ashaba Katabazi, Edgar Kigozi, Moses L. Joloba, David Patrick Kateete
Scientific Reports.2023;[Epub] CrossRef - Molecular definition of severe acute respiratory syndrome coronavirus 2 receptor‐binding domain mutations: Receptor affinity versus neutralization of receptor interaction
Monique Vogel, Gilles Augusto, Xinyue Chang, Xuelan Liu, Daniel Speiser, Mona O. Mohsen, Martin F. Bachmann
Allergy.2022; 77(1): 143. CrossRef - Peptides and peptidomimetics as therapeutic agents for Covid‐19
Achyut Dahal, Jafrin Jobayer Sonju, Konstantin G. Kousoulas, Seetharama D. Jois
Peptide Science.2022;[Epub] CrossRef - Emergence of unique SARS-CoV-2 ORF10 variants and their impact on protein structure and function
Sk. Sarif Hassan, Kenneth Lundstrom, Ángel Serrano-Aroca, Parise Adadi, Alaa A.A. Aljabali, Elrashdy M. Redwan, Amos Lal, Ramesh Kandimalla, Tarek Mohamed Abd El-Aziz, Pabitra Pal Choudhury, Gajendra Kumar Azad, Samendra P. Sherchan, Gaurav Chauhan, Murta
International Journal of Biological Macromolecules.2022; 194: 128. CrossRef - COVID-19: comprehensive review on mutations and current vaccines
Ananda Vardhan Hebbani, Swetha Pulakuntla, Padmavathi Pannuru, Sreelatha Aramgam, Kameswara Rao Badri, Vaddi Damodara Reddy
Archives of Microbiology.2022;[Epub] CrossRef - Prediction of the Effects of Nonsynonymous Variants on SARS-CoV-2 Proteins
Boon Zhan Sia, Wan Xin Boon, Yoke Yee Yap, Shalini Kumar, Chong Han Ng
F1000Research.2022; 11: 9. CrossRef - Identification of SARS-CoV-2 Variants and Their Clinical Significance in Hefei, China
Xiao-wen Cheng, Jie Li, Lu Zhang, Wen-jun Hu, Lu Zong, Xiang Xu, Jin-ping Qiao, Mei-juan Zheng, Xi-wen Jiang, Zhi-kun Liang, Yi-fan Zhou, Ning Zhang, Hua-qing Zhu, Yuan-hong Xu
Frontiers in Medicine.2022;[Epub] CrossRef - Promising inhibitors against main protease of SARS CoV-2 from medicinal plants: In silico identification
OLUWAKEMI EBENEZER, MICHAEL SHAPI
Acta Pharmaceutica.2022; 72(2): 159. CrossRef - Genomic surveillance of SARS-CoV-2 in the state of Delaware reveals tremendous genomic diversity
Karl R. Franke, Robert Isett, Alan Robbins, Carrie Paquette-Straub, Craig A. Shapiro, Mary M. Lee, Erin L. Crowgey, Pierre Roques
PLOS ONE.2022; 17(1): e0262573. CrossRef - The importance of accessory protein variants in the pathogenicity of SARS-CoV-2
Sk. Sarif Hassan, Pabitra Pal Choudhury, Guy W. Dayhoff, Alaa A.A. Aljabali, Bruce D. Uhal, Kenneth Lundstrom, Nima Rezaei, Damiano Pizzol, Parise Adadi, Amos Lal, Antonio Soares, Tarek Mohamed Abd El-Aziz, Adam M. Brufsky, Gajendra Kumar Azad, Samendra P
Archives of Biochemistry and Biophysics.2022; 717: 109124. CrossRef - The mutational dynamics of the SARS-CoV-2 virus in serial passages in vitro
Sissy Therese Sonnleitner, Stefanie Sonnleitner, Eva Hinterbichler, Hannah Halbfurter, Dominik B.C. Kopecky, Stephan Koblmüller, Christian Sturmbauer, Wilfried Posch, Gernot Walder
Virologica Sinica.2022; 37(2): 198. CrossRef - Evolutionary dynamics of SARS-CoV-2 circulating in Yogyakarta and Central Java, Indonesia: sequence analysis covering furin cleavage site (FCS) region of the spike protein
Nastiti Wijayanti, Faris Muhammad Gazali, Endah Supriyati, Mohamad Saifudin Hakim, Eggi Arguni, Marselinus Edwin Widyanto Daniwijaya, Titik Nuryastuti, Matin Nuhamunada, Rahma Nabilla, Sofia Mubarika Haryana, Tri Wibawa
International Microbiology.2022; 25(3): 531. CrossRef - Prediction of the effects of the top 10 nonsynonymous variants from 30229 SARS-CoV-2 strains on their proteins
Boon Zhan Sia, Wan Xin Boon, Yoke Yee Yap, Shalini Kumar, Chong Han Ng
F1000Research.2022; 11: 9. CrossRef - The Mutational Landscape of SARS-CoV-2 Variants of Concern Recovered From Egyptian Patients in 2021
Mohamed G. Seadawy, Reem Binsuwaidan, Badriyah Alotaibi, Thanaa A. El-Masry, Bassem E. El-Harty, Ahmed F. Gad, Walid F. Elkhatib, Maisra M. El-Bouseary
Frontiers in Microbiology.2022;[Epub] CrossRef - Genomic surveillance, evolution and global transmission of SARS-CoV-2 during 2019–2022
Nadim Sharif, Khalid J. Alzahrani, Shamsun Nahar Ahmed, Afsana Khan, Hamsa Jameel Banjer, Fuad M. Alzahrani, Anowar Khasru Parvez, Shuvra Kanti Dey, Jayanta Bhattacharya
PLOS ONE.2022; 17(8): e0271074. CrossRef - Comparison of Intracellular Transcriptional Response of NHBE Cells to Infection with SARS-CoV-2 Washington and New York Strains
Tiana M. Scott, Antonio Solis-Leal, J. Brandon Lopez, Richard A. Robison, Bradford K. Berges, Brett E. Pickett
Frontiers in Cellular and Infection Microbiology.2022;[Epub] CrossRef - The Delta and Omicron Variants of SARS-CoV-2: What We Know So Far
Vivek Chavda, Rajashri Bezbaruah, Kangkan Deka, Lawandashisha Nongrang, Tutumoni Kalita
Vaccines.2022; 10(11): 1926. CrossRef - SARS-CoV-2’NİN SÜREGELEN EVRİMİ: PANDEMİNİN SONUNA NE KADAR YAKINIZ?
Elmas Pınar KAHRAMAN KILBAŞ, Mustafa ALTINDİŞ
Journal of Biotechnology and Strategic Health Rese.2022; 6(3): 201. CrossRef - Complete Genomic Characterisation and Mutation Patterns of Iraqi SARS-CoV-2 Isolates
Jivan Qasim Ahmed, Sazan Qadir Maulud
Diagnostics.2022; 13(1): 8. CrossRef - Comprehensive annotations of the mutational spectra of SARS‐CoV‐2 spike protein: a fast and accurate pipeline
Mohammad Shaminur Rahman, Mohammad Rafiul Islam, Mohammad Nazmul Hoque, Abu Sayed Mohammad Rubayet Ul Alam, Masuda Akther, Joynob Akter Puspo, Salma Akter, Azraf Anwar, Munawar Sultana, Mohammad Anwar Hossain
Transboundary and Emerging Diseases.2021; 68(3): 1625. CrossRef - Genomic and proteomic mutation landscapes of SARS‐CoV‐2
Christian Luke D. C. Badua, Karol Ann T. Baldo, Paul Mark B. Medina
Journal of Medical Virology.2021; 93(3): 1702. CrossRef - Variant analysis of the first Lebanese SARS-CoV-2 isolates
Mhamad Abou-Hamdan, Kassem Hamze, Ali Abdel Sater, Haidar Akl, Nabil El-zein, Israa Dandache, Fadi Abdel-sater
Genomics.2021; 113(1): 892. CrossRef - Global SNP analysis of 11,183 SARS‐CoV‐2 strains reveals high genetic diversity
Fangfeng Yuan, Liping Wang, Ying Fang, Leyi Wang
Transboundary and Emerging Diseases.2021; 68(6): 3288. CrossRef - SARS-CoV-2 hot-spot mutations are significantly enriched within inverted repeats and CpG island loci
Pratik Goswami, Martin Bartas, Matej Lexa, Natália Bohálová, Adriana Volná, Jiří Červeň, Veronika Červeňová, Petr Pečinka, Vladimír Špunda, Miroslav Fojta, Václav Brázda
Briefings in Bioinformatics.2021; 22(2): 1338. CrossRef - Deciphering the Subtype Differentiation History of SARS-CoV-2 Based on a New Breadth-First Searching Optimized Alignment Method Over a Global Data Set of 24,768 Sequences
Qianyu Lin, Yunchuanxiang Huang, Ziyi Jiang, Feng Wu, Lan Ma
Frontiers in Genetics.2021;[Epub] CrossRef - Temporal increase in D614G mutation of SARS-CoV-2 in the Middle East and North Africa
Malik Sallam, Nidaa A. Ababneh, Deema Dababseh, Faris G. Bakri, Azmi Mahafzah
Heliyon.2021; 7(1): e06035. CrossRef - Environmental aspect and applications of nanotechnology to eliminate COVID-19 epidemiology risk
Eman Serag, Marwa El-Zeftawy
Nanotechnology for Environmental Engineering.2021;[Epub] CrossRef - Genomic Characterization and Phylogenetic Analysis of SARS-CoV-2 in Libya
Silvia Fillo, Francesco Giordani, Anella Monte, Giovanni Faggioni, Riccardo De Santis, Nino D’Amore, Stefano Palomba, Taher Hamdani, Kamel Taloa, Atef Belkhir Jumaa, Siraj Bitrou, Ahmed Alaruusi, Wadie Mad, Abdulaziz Zorgani, Omar Elahmer, Badereddin Anna
Microbiology Research.2021; 12(1): 138. CrossRef - SARS-CoV-2 Entry Related Viral and Host Genetic Variations: Implications on COVID-19 Severity, Immune Escape, and Infectivity
Szu-Wei Huang, Sheng-Fan Wang
International Journal of Molecular Sciences.2021; 22(6): 3060. CrossRef - Emergence of novel SARS-CoV-2 variants in the Netherlands
Aysun Urhan, Thomas Abeel
Scientific Reports.2021;[Epub] CrossRef - Lung organoid simulations for modelling and predicting the effect of mutations on SARS-CoV-2 infectivity
Sally Esmail, Wayne R. Danter
Computational and Structural Biotechnology Journal.2021; 19: 1701. CrossRef - MSC-derived exosomes carrying a cocktail of exogenous interfering RNAs an unprecedented therapy in era of COVID-19 outbreak
Monire Jamalkhah, Yasaman Asaadi, Mohammadreza Azangou-Khyavy, Javad Khanali, Masoud Soleimani, Jafar Kiani, Ehsan Arefian
Journal of Translational Medicine.2021;[Epub] CrossRef - Mutations and Epidemiology of SARS-CoV-2 Compared to Selected Corona Viruses during the First Six Months of the COVID-19 Pandemic: A Review
Mirriam M. Nzivo, Nancy L.M. Budambula
Journal of Pure and Applied Microbiology.2021; 15(2): 524. CrossRef - The First Molecular Characterization of Serbian SARS-CoV-2 Isolates From a Unique Early Second Wave in Europe
Danijela Miljanovic, Ognjen Milicevic, Ana Loncar, Dzihan Abazovic, Dragana Despot, Ana Banko
Frontiers in Microbiology.2021;[Epub] CrossRef - Identification of a High-Frequency Intrahost SARS-CoV-2 Spike Variant with Enhanced Cytopathic and Fusogenic Effects
Lynda Rocheleau, Geneviève Laroche, Kathy Fu, Corina M. Stewart, Abdulhamid O. Mohamud, Marceline Côté, Patrick M. Giguère, Marc-André Langlois, Martin Pelchat, Dimitrios Paraskevis
mBio.2021;[Epub] CrossRef - Notable and Emerging Variants of SARS-CoV-2 Virus: A Quick Glance
Sagar Dholariya, Deepak Narayan Parchwani, Ragini Singh, Amit Sonagra, Anita Motiani, Digishaben Patel
Indian Journal of Clinical Biochemistry.2021; 36(4): 451. CrossRef - Genomic characterization of SARS‐CoV‐2 isolates from patients in Turkey reveals the presence of novel mutations in spike and nsp12 proteins
Erdem Sahin, Gulendam Bozdayi, Selin Yigit, Hager Muftah, Murat Dizbay, Ozlem G. Tunccan, Isil Fidan, Kayhan Caglar
Journal of Medical Virology.2021; 93(10): 6016. CrossRef - Temporal landscape of mutational frequencies in SARS-CoV-2 genomes of Bangladesh: possible implications from the ongoing outbreak in Bangladesh
Otun Saha, Israt Islam, Rokaiya Nurani Shatadru, Nadira Naznin Rakhi, Md. Shahadat Hossain, Md. Mizanur Rahaman
Virus Genes.2021; 57(5): 413. CrossRef - SARS-CoV-2 receptor-binding mutations and antibody contact sites
Marios Mejdani, Kiandokht Haddadi, Chester Pham, Radhakrishnan Mahadevan
Antibody Therapeutics.2021; 4(3): 149. CrossRef - Evolutionary trajectory of SARS-CoV-2 and emerging variants
Jalen Singh, Pranav Pandit, Andrew G. McArthur, Arinjay Banerjee, Karen Mossman
Virology Journal.2021;[Epub] CrossRef - Molecular characterization of SARS-CoV-2 from Bangladesh: implications in genetic diversity, possible origin of the virus, and functional significance of the mutations
Md. Marufur Rahman, Shirmin Bintay Kader, S.M. Shahriar Rizvi
Heliyon.2021; 7(8): e07866. CrossRef - Exploring the Binding Mechanism of PF-07321332 SARS-CoV-2 Protease Inhibitor through Molecular Dynamics and Binding Free Energy Simulations
Bilal Ahmad, Maria Batool, Qurat ul Ain, Moon Suk Kim, Sangdun Choi
International Journal of Molecular Sciences.2021; 22(17): 9124. CrossRef - Characterization of the SARS-CoV-2 genomes in Egypt in first and second waves of infection
Abdel-Rahman N. Zekri, Abeer A. Bahnasy, Mohamed M. Hafez, Zeinab K. Hassan, Ola S. Ahmed, Hany K. Soliman, Enas R. El-Sisi, Mona H. Salah El Dine, May S. Solimane, Lamyaa S. Abdel Latife, Mohamed G. Seadawy, Ahmed S. Elsafty, Mohamed Abouelhoda
Scientific Reports.2021;[Epub] CrossRef - A Global Mutational Profile of SARS-CoV-2: A Systematic Review and Meta-Analysis of 368,316 COVID-19 Patients
Wardah Yusof, Ahmad Adebayo Irekeola, Yusuf Wada, Engku Nur Syafirah Engku Abd Rahman, Naveed Ahmed, Nurfadhlina Musa, Muhammad Fazli Khalid, Zaidah Abdul Rahman, Rosline Hassan, Nik Yusnoraini Yusof, Chan Yean Yean
Life.2021; 11(11): 1224. CrossRef - Phylogenetic and full-length genome mutation analysis of SARS-CoV-2 in Indonesia prior to COVID-19 vaccination program in 2021
Reviany V. Nidom, Setyarina Indrasari, Irine Normalina, Astria N. Nidom, Balqis Afifah, Lestari Dewi, Andra K. Putra, Arif N. M. Ansori, Muhammad K. J. Kusala, Mohammad Y. Alamudi, Chairul A. Nidom
Bulletin of the National Research Centre.2021;[Epub] CrossRef - Global Pandemic as a Result of Severe Acute Respiratory Syndrome Coronavirus 2 Outbreak: A Biomedical Perspective
Charles Arvind Sethuraman Vairavan, Devarani Rameshnathan, Nagaraja Suryadevara, Gnanendra Shanmugam
Journal of Pure and Applied Microbiology.2021; 15(4): 1759. CrossRef - Predicting the Molecular Mechanism of Sini Jia Renshen Decoction in Treating Severe COVID-19 Patients Based on Network Pharmacology and Molecular Docking
Yi Wen Liu, Ai Xia Yang, Li Lu, Tie Hua Huang
Natural Product Communications.2021; 16(12): 1934578X2110592. CrossRef - Can SARS-CoV-2 Accumulate Mutations in the S-Protein to Increase Pathogenicity?
Aditya K. Padhi, Timir Tripathi
ACS Pharmacology & Translational Science.2020; 3(5): 1023. CrossRef - Optimized Pseudotyping Conditions for the SARS-COV-2 Spike Glycoprotein
Marc C. Johnson, Terri D. Lyddon, Reinier Suarez, Braxton Salcedo, Mary LePique, Maddie Graham, Clifton Ricana, Carolyn Robinson, Detlef G. Ritter, Viviana Simon
Journal of Virology.2020;[Epub] CrossRef - A Crowned Killer’s Résumé: Genome, Structure, Receptors, and Origin of SARS-CoV-2
Shichuan Wang, Mirko Trilling, Kathrin Sutter, Ulf Dittmer, Mengji Lu, Xin Zheng, Dongliang Yang, Jia Liu
Virologica Sinica.2020; 35(6): 673. CrossRef - Host or pathogen-related factors in COVID-19 severity?
Christian Gortázar, Francisco J Rodríguez del-Río, Lucas Domínguez, José de la Fuente
The Lancet.2020; 396(10260): 1396. CrossRef - Host or pathogen-related factors in COVID-19 severity? – Authors' reply
Lucy C Okell, Robert Verity, Aris Katzourakis, Erik M Volz, Oliver J Watson, Swapnil Mishra, Patrick Walker, Charlie Whittaker, Christl A Donnelly, Steven Riley, Azra C Ghani, Axel Gandy, Seth Flaxman, Neil M Ferguson, Samir Bhatt
The Lancet.2020; 396(10260): 1397. CrossRef - Recent updates on COVID-19: A holistic review
Shweta Jakhmola, Omkar Indari, Dharmendra Kashyap, Nidhi Varshney, Annu Rani, Charu Sonkar, Budhadev Baral, Sayantani Chatterjee, Ayan Das, Rajesh Kumar, Hem Chandra Jha
Heliyon.2020; 6(12): e05706. CrossRef - Impact of Genetic Variability in ACE2 Expression on the Evolutionary Dynamics of SARS-CoV-2 Spike D614G Mutation
Szu-Wei Huang, Sorin O. Miller, Chia-Hung Yen, Sheng-Fan Wang
Genes.2020; 12(1): 16. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite