Articles
- Page Path
- HOME > Osong Public Health Res Perspect > Volume 13(5); 2022 > Article
-
Original Article
Investigation of SARS-CoV-2 lineages and mutations circulating in a university-affiliated hospital in South Korea analyzed using Oxford Nanopore MinION sequencing -
Hyaekang Kim1
 , Sung Hee Chung1, Hyun Soo Kim2, Han-Sung Kim3, Wonkeun Song4, Ki Ho Hong5, Jae-Seok Kim1
, Sung Hee Chung1, Hyun Soo Kim2, Han-Sung Kim3, Wonkeun Song4, Ki Ho Hong5, Jae-Seok Kim1 -
Osong Public Health and Research Perspectives 2022;13(5):360-369.
DOI: https://doi.org/10.24171/j.phrp.2022.0183
Published online: October 11, 2022
1Department of Laboratory Medicine, Kangdong Sacred Heart Hospital, Hallym University College of Medicine, Seoul, Korea
2Department of Laboratory Medicine, Hallym University Dongtan Sacred Heart Hospital, Hwaseong, Korea
3Department of Laboratory Medicine, Hallym University Sacred Heart Hospital, Anyang, Korea
4Department of Laboratory Medicine, Hallym University Kangnam Sacred Heart Hospital, Seoul, Korea
5Department of Laboratory Medicine, Yonsei University College of Medicine, Seoul, Korea
- Corresponding author: Jae-Seok Kim Department of Laboratory Medicine, Kangdong Sacred Heart Hospital, Hallym University College of Medicine, 150 Seongan-ro, Gangdong-gu, Seoul 05355, Korea E-mail: jaeseok@kdh.or.kr
© 2022 Korea Disease Control and Prevention Agency.
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Abstract
-
Objectives
- Despite the introduction of vaccines, treatments, and massive diagnostic testing, the evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has continued to overcome barriers that had slowed its previous spread. As the virus evolves towards increasing fitness, it is critical to continue monitoring the occurrence of new mutations that could evade human efforts to control them.
-
Methods
- We performed whole-genome sequencing using Oxford Nanopore MinION sequencing on 58 SARS-CoV-2 isolates collected during the ongoing coronavirus disease 2019 pandemic at a tertiary hospital in South Korea and tracked the emergence of mutations responsible for massive spikes in South Korea.
-
Results
- The differences among lineages were more pronounced in the spike gene, especially in the receptor-binding domain (RBD), than in other genes. Those RBD mutations could compromise neutralization by antibodies elicited by vaccination or previous infections. We also reported multiple incidences of Omicron variants carrying mutations that could impair the diagnostic sensitivity of reverse transcription-polymerase chain reaction-based testing.
-
Conclusion
- These results provide an understanding of the temporal changes of variants and mutations that have been circulating in South Korea and their potential impacts on antigenicity, therapeutics, and diagnostic escape of the virus. We also showed that the utilization of the nanopore sequencing platform and the ARTIC workflow can provide convenient and accurate SARS-CoV-2 genomic surveillance even at a single hospital.
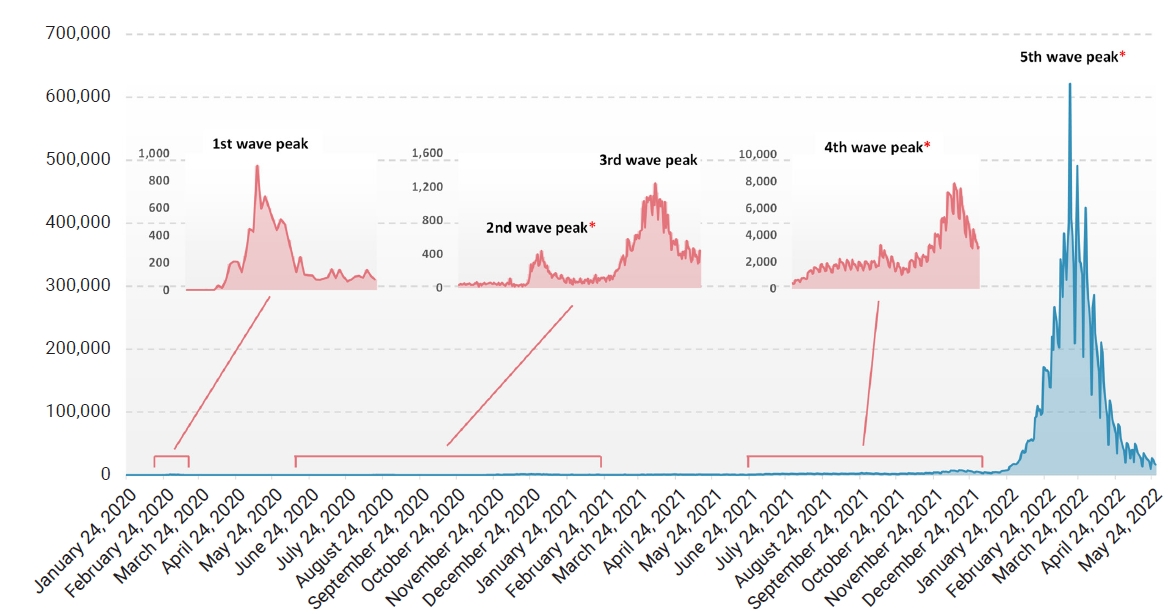
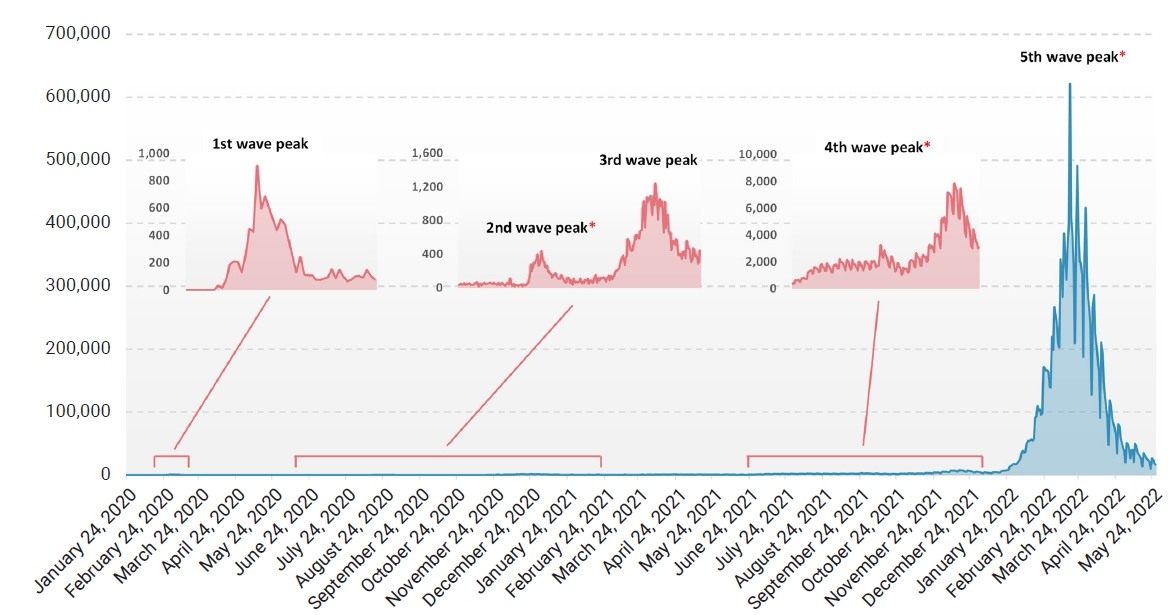
- Since its first emergence in December 2019, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has rapidly spread globally, and 4 months later the World Health Organization officially declared it a pandemic [1]. Since then, over 500 million human cases, including over 6 million deaths, have been reported worldwide so far, yielding several variants of interest (VOIs) and variants of concern (VOCs). The first case in South Korea was reported in January 2020, followed by a rapid increase in reported cases, which led to the first wave peak in February 2020 [2]. There have been several rises in the number of new infections in South Korea [2], including the latest surge of Omicron subvariants. Despite the strict restrictions on human activity and the introduction of vaccines and treatments, the evolution of the virus has continued to overcome barriers that had previously slowed its spread. The highest number of infections caused by new emerging variants has always exceeded the previous high, and the recent Omicron peak reached about 80 times higher than the previous Delta peak in South Korea [3].
- During the course of the pandemic, more than 10 billion genome sequences of SARS-CoV-2 have been gathered worldwide, leading to the discovery that its continuous evolution is the fundamental driving force behind the prolonged pandemic [4]. Several genomic surveillance studies have also revealed that the accumulation of mutations has been heterogeneous along the genome and multiple genetic sites, such as the genes coding for the S and N proteins, were under positive selection [5,6]. As the virus evolves towards increasing fitness, it is necessary to continue monitoring the occurrence of new mutations that could increase infectivity or transmission efficiency, especially in response to these human efforts to control them.
- In this study, we performed whole-genome sequencing using Oxford Nanopore MinION on 58 SARS-CoV-2 isolates collected during the ongoing coronavirus disease 2019 (COVID-19) pandemic (the 2nd, 4th, and 5th waves of infections) at a university-affiliated hospital in South Korea and tracked the emergence of mutations of certain variants responsible for the massive spikes in South Korea. Our results provide an understanding of the temporal change of variants and mutations that have been circulating in South Korea and the potential contribution of the evolving mutations on antigenicity, therapeutics, and diagnostic escape of the virus. We were also able to demonstrate that the utilization of the nanopore sequencing platform and the ARTIC workflow could provide convenient and accurate SARS-CoV-2 genomic surveillance even at a single hospital.
Introduction
- Sample Collection and Diagnostics
- Nasopharyngeal and oral swab specimens from suspected cases were collected as part of routine surveillance at Kangdong Sacred Heart Hospital, Seoul, South Korea. RNA was extracted using a STARMag Universal Cartridge Kit (Seegene Inc., Seoul, Korea), and real-time reverse-transcription polymerase chain reaction (RT-PCR) with Seegene Allplex SARS-CoV-2 assay (Seegene Inc.) on a Bio-Rad CFX96 thermal cycler (Bio-Rad Laboratories, Hercules, CA, USA) was performed. Among samples that tested positive, those collected from June 2020 to September 2020 (Wuhan-type epidemic), November 2021 to December 2021 (Delta epidemic), and February 2022 to April 2022 (Omicron epidemic), which coincided with the 2nd, 4th, and 5th waves of the pandemic in South Korea, were subjected to whole-genome sequencing (Figure 1; Table S1).
- Amplicon-Based Whole-Genome Sequencing
- We performed whole-genome sequencing on 58 SARS-CoV-2 isolates following the ARTIC nCoV-2019 sequencing protocol. The cDNA synthesis and PCR amplification were performed according to the ARTIC amplicon sequencing protocol, using cDNA synthesis Automix (Seegene Inc.) in combination with the ARTIC nCoV-2019 V3 amplicon set. The PCR products were treated with a ligation kit (SQK-LSK109; Oxford Nanopore Technologies, Oxford, UK) and a barcoding kit (EXP-SFB001; Oxford Nanopore Technologies). Libraries were sequenced with the Oxford Nanopore MinION device using ONT R9.4.1 flow cells (Oxford Nanopore Technologies) for 12 hours.
- Generation of Consensus Genomes and Mutational Profiling
- For the construction of consensus sequences, the recommended ARTIC bioinformatics workflow (https://github.com/artic-network/fieldbioinformatics) was used. Generated sequences were manually assessed to verify the mutations called by the ARTIC pipeline. The genomes were classified into PANGO lineages using Pangolin v.4.0.6 [7], and mutation annotation was performed using Nextclade (https://clades.nextstrain.org) and Coronapp (http://giorgilab.unibo.it/coronannotator/). The genome sequences used for comparison were retrieved from the GISAID database (https://www.gisaid.org/) on April 6, 2022. Sequences were aligned using ClustalW 1.2.1 [8], and a maximum-likelihood phylogenetic tree was constructed using FastTree 2.1 [9] with 1,000 bootstrap replications. The Wuhan-Hu-1 sequence (RefSeq: NC_045512.2) was used as a reference genome and for rooting the tree as an outgroup. The tree was visualized using Interactive Tree of Life ver. 5 (https://itol.embl.de). Antibody escape was calculated using an escape calculator for the SARS-CoV-2 receptor-binding domain (RBD) [10].
- Ethics Approval
- This study was approved by the Institutional Review Board (IRB) of Kangdong Sacred Heart Hospital, Seoul, South Korea (IRB No: NON2021-001-001) and performed in accordance with the principles of the Declaration of Helsinki.
Materials and Methods
- Temporal Changes of SARS-CoV-2 Variants in South Korea
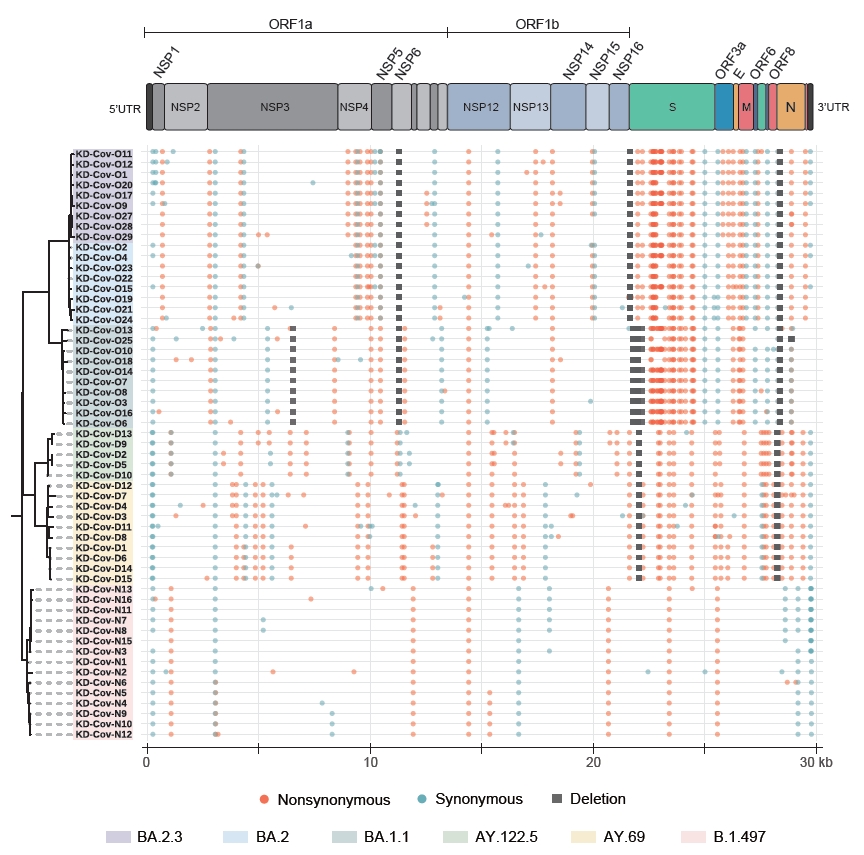
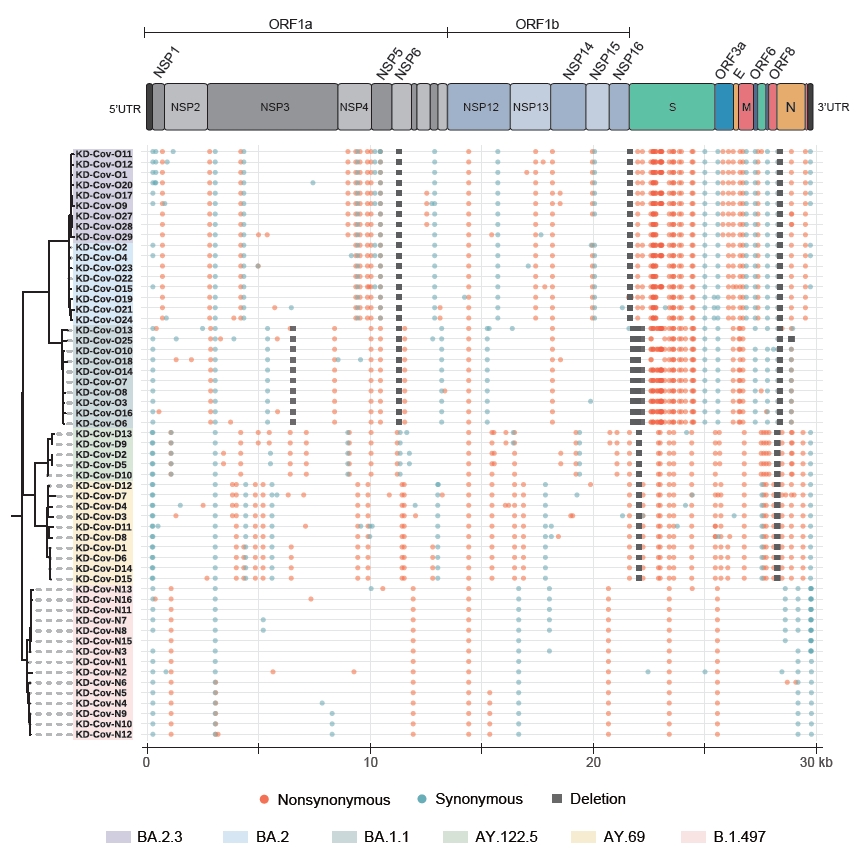
- The ARTIC protocol generated 250,686±45,498 raw reads, totaling approximately 109±19 Mbp of sequences for each sample. The mean read qualities ranged between 11.6 and 13.9 (Table S1). All 58 sequenced samples showed at least 85% of the genome covered at least 20-fold, and 48 samples had more than 95% coverage (Table S2). Fifty-seven samples with at least 90% coverage were used in the subsequent whole-genome comparison (Table S1). In total, 6 SARS-CoV-2 lineages were identified in the 58 samples based on Pangolin [7] and Nextclade [11]. The samples collected between June 2020 and September 2020 (second wave of infections) were assigned to the B.1.497 (20C) lineage, and the samples collected between November 2021 and December 2021 (4th wave) belonged to the Delta VOC, which consisted of AY.69 (21I) and AY.122.5 (21J) (Table S1). Twenty-eight samples collected between March 2022 and April 2022 (5th wave) belonged to the Omicron VOC, including 11, 8, and 9 classified as BA.1.1 (21K), BA.2 (21L), and BA.2.3 (21L), respectively. The BA.2 and BA.2.3 lineages are currently the major causes of ongoing cases in South Korea [3]. The B.1.497 lineage had an average of 14.7 mutations, and the number of synonymous and non-synonymous mutations was similar on average (Figure S1; Table S3). However, an average of 45.1 and 44.4 mutations were identified in the 2 Delta subvariants, and the number of non-synonymous mutations was more than twice greater than that of synonymous mutations (Figure S1; Table S3). For the 3 Omicron subvariants (8 intact BA.1.1, 3 BA.2, and 5 BA.2.3 sequences were considered), the average numbers of mutations were 61.9, 66.7, and 70 for BA.1.1, BA.2, and BA.2.3 respectively, and the differences among them were also mainly due to the accumulation of non-synonymous mutations (42.1, 47, and 49.4 on average) (Figure S1; Table S3). The differences in the accumulation of non-synonymous and synonymous mutations were greater in the S region, compared to the other gene regions (Figure S1). As the virus evolved, while the number of synonymous mutations maintained below 7 in the S gene, non-synonymous mutations markedly increased to an average of 28, 22, and 23 in BA.1.1, BA.2, and BA.2.3, respectively (Figure S1).
- The divergence between lineages was the greatest in the spike protein. The BA.1.1 subvariant contained about 32 new mutations in the spike encoding region compared to the Delta variants, and the BA.2 and BA.2.3 subvariants gained about 9 new mutations (T19I, A27S, G142D, S371F, V213G, T376A, D405N, R408S, and 24–26 deletion) compared to BA.1.1 (Tables S2, S3). Meanwhile, the 2 subvariants lacked 14 mutations, including 69–70 del, 142–144 del, and 211 del. Although most of the amino acid changes in the spike were fixed at the individual level, KD-Cov-O19 carried 2 unique mutations, L5F and H49Y (Tables S2, S3).
- Impacts of the Emergence of Mutations on Infectivity and Therapeutics
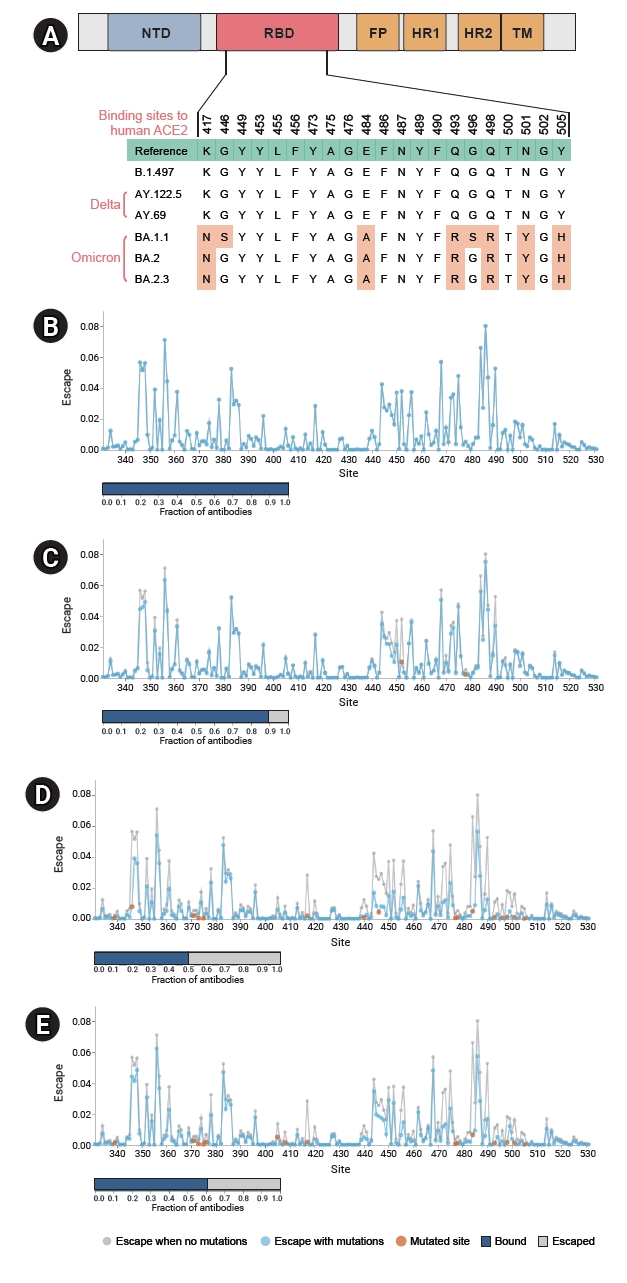
- As evident from Figure 2 and Table S4, the differences among lineages were more pronounced in the spike gene. While the B.1.497 possessed only 1 fixed mutation in the gene, 6–7 and 27–31 fixed mutations were identified in genomes of the Delta (AY.122.5 and AY.69) and the Omicron (BA.1.1, BA.2, and BA.2.3) variants, respectively. Markedly, the majority of the mutations gained by the Omicron variants were located in the RBD region of the gene, which had 7.5–8 times more substitutions (16 for BA.1.1 and 15 for BA.2 and BA.2.3) than the Delta subvariants in the region, and all these substitutions were non-synonymous changes. Of the 16 changes in Omicron BA.1.1 and 15 changes in the Omicron BA.2 and BA.2.3 subvariants in the RBD, 8 (K417N, G446S, E484A, Q493R, G496S, Q498R, N501Y, and Y505H) and 6 (K417N, E484A, Q493R, Q498R, N501Y, and Y505H), respectively, mapped to the positions proposed to be critical for binding to human ACE2 [12], and the residues remained unchanged in other lineages (Figure 3A).
- The antibody-escape calculator [10] estimated that the combination of the RBD mutations of the Delta subvariants reduced the fraction of binding antibodies to below 0.9 (Figure 3C). The mutations that emerged in the Omicron variants could lead to more immune escape by dropping the fraction to 0.5 (BA.1.1) and 0.6 (BA.2 and BA.2.3), and the difference between these subvariants was induced by additional R346K, N440K, G446S, and G496S mutations in the BA.1.1 lineage (Figure 3D, E).
- In the Mpro coding region, which is an important antiviral drug target of Paxlovid (Pfizer) for treatment of the virus, 1 B.1.497 isolate (KD-Cov-N13) was observed to have a change in the 168 position, and 2 AY.69 isolates, D7 and D11, harbored M264I and a synonymous change in the 10078 nucleotide position, respectively (Table S3). The BA.1.1 lineage carried 1 mutation (P132H), and 4 types of substitution (synonymous changes in the 10198, 10432, and 10447 nucleotide positions and P132H) were observed in BA.2 and BA.2.3. The substitution at the 10432 position occurred only in KD-Cov-O11, BA.2.3, which had the most substitutions in the nsp5 region among our isolates.
- The Effect of the Emergence of Mutations on Diagnostic Sensitivity
- Routine diagnostic testing has been performed at our diagnostic facility since the beginning of the SARS-CoV-2 pandemic. However, since February 2022, we have observed a few cases (KD-CoV-25, 26, 27, 28, and 29) showing discrepant results in target gene detection with a commercial assay (Allplex SARS-CoV-2 assay) which targets N, E, and RNA-dependent RNA polymerase (RdRP)/S gene fragments. KD-Cov-O25 and O26, a mother-child pair, showed N gene drop-out, despite having cycle threshold (Ct) values below 25 for both the E and RdRP/S genes (Table 1). In the KD-Cov-O27 and O28 samples, another mother-child pair, the Ct values for N and the other 2 (E and RdRP/S) differed by about 5 cycles, and regarding KD-Cov-O29, the Ct value for RdRP/S was 8 cycles higher than those of the E and N genes (Table 1). Deletions were identified in position 28877-28894, leading to the deletion of 6 amino acids (203–208 del) in the N protein of the KD-CoV-O25 and O26 pair. The KD-CoV-O27 and O28 pair carried an A208V mutation (C28896T) in the protein, overlapping the deleted region of the above-mentioned isolates. The only change observed in the KD-Cov-O29 genome was an additional M657T mutation (T15437C) in NSP12 compared to BA.2.3 isolates with normal Ct values. Although information on the primers and probes used in commercial SARS-CoV-2 assays is not generally disclosed, the mutations observed in the above cases may affect the RT-PCR diagnostic efficacy of the Allplex SARS-CoV-2 assay. To date, a total of 1,281 cases with deletions in the 203-208 position were reported on GISAID (10,140,904 deposited N gene sequences as of April 6, 2022). Interestingly, this deletion has been sporadically occurring in Canada, the USA, Brazil, Germany, Italy, and several other countries since its first appearing on GISAID in early 2020 (EPI_ISL_3717678, Isolation: India, March 19, 2020) before the emergence of Omicron. Furthermore, it was not specific to the Omicron variant and was also observed in Alpha, Delta, Gamma, Omicron, and other lineages. It was most prevalent in the USA and Brazil (397 and 186 instances, respectively). However, in South Korea, the deletion was first observed in January 2022 in the genome sequence of the Omicron BA.1.1 variant on GISAID and has maintained a low frequency (7 instances/11,785 Omicron sequences). It has also been exclusively observed in the Omicron subvariant (BA.1.1) so far in South Korea. The A208V mutation has been observed in 4,447 genome sequences, and the majority of them originated from the USA (1,009) and England (897).
Results
- In this study, we performed whole-genome sequencing on 58 SARS-CoV-2 isolates collected during the ongoing COVID-19 pandemic at a single hospital to track the emergence of mutations of certain variants responsible for the massive spikes in South Korea. The differences among lineages were more pronounced in the spike gene, especially in the RBD region. Since changes in the RBD region may substantially change the antigenicity and susceptibility to pre-existing antibodies [12,13], mutations of emerging viruses in this region should be closely monitored. The new mutations gained by the Omicron variants were mostly located in the critical binding sites to human ACE2. Interestingly, G446S and G496S were exclusively detected in BA.1.1. Another study reported that G496 was the residue introducing the most significant effects on the RBD-ACE2 interaction [14], and the G496S mutation was also suggested to reinforce or generate chemical bonding at the interface, along with G446S and N501Y [15]. Of the mutually detected mutations in the 3 Omicron subvariants, a previous study showed that Q493R and N501Y, along with S477N, formed a new interaction with human ACE2, which enhanced the binding of the Omicron variant to human ACE2 [16]. The Y505H mutation did not seem to have an impact on binding [17].
- We then analyzed how the emerging RBD mutations could compromise neutralization by antibodies elicited by vaccination or natural infection, using a recently developed escape calculator tool that utilized a deep mutational scanning approach. Previous studies demonstrated that the calculator correlated well with experimentally measured neutralization titers [10]. The results indicated that the combination of RBD mutations that emerged in the Omicron variants could lead to more immune escape compared to the previous lineages. The difference between the Omicron subvariants was induced by additional R346K, N440K, G446S, and G496S mutations in the BA.1.1 lineage (Figure 3D, E). Motozono et al. [18] showed that Omicron BA.1 with the G446S mutation, located adjacent to NF9 epitope, showed greater inhibition of replication by vaccine-induced T cells as a result of enhanced presentation and recognition of the antigenic peptide on the spike protein. As shown in Figure 3B–E, the mutation at position 484 was among the largest drivers of antigenic changes [19]. In a study using 19 monoclonal antibodies, substitutions at this position occurred the most frequently [20], and all amino acid changes to A, K, Q, P, D, and G reduced recognition by antibodies [20,21]. The E484A mutation detected in Omicron variants also ablated neutralization by bamlanivimab, a therapeutic monoclonal antibody [22]. The E484K mutation carried in Beta, Gamma, Zeta, Eta, and Theta variants [23] is also well known for its capability of reducing antibody binding, and it emerged in a patient during the use of monoclonal antibodies or convalescent plasma therapy [24].
- Rapid emergence of new variants with higher mutation loads also raises concern for the clinical effectiveness of currently used treatments. The non-monoclonal antibody therapeutics currently approved and used to treat SARS-CoV-2 are nirmatrelvir (an antiviral component of Paxlovid), remdesivir, and molnupiravir. Nirmatrelvir inhibits Mpro, and both remdesivir and molnupiravir are RdRP inhibitors. According to previous studies, there are 26 binding site amino acid residues in NSP5 with nirmatrelvir, and key interaction sites were discovered [25,26]. Interestingly, the P168S mutation identified only in KD-Cov-N13 was located at this site, but the sample was collected in September 2020, which indicates that the mutation was not driven by selective drug pressure. Although other substitutions were not located in the direct binding site, the synonymous change in the 10198 nucleotide position, frequently detected in lineages BA.2 and BA.2.3, was located in the proximity of the S2 binding pocket of nirmatrelvir [25]. P132H, one of the most prevalent NSP5 variants, was also detected in all the Omicron isolates in this study, but several studies have shown that it is unlikely to affect nirmatrelvir binding affinity, as the residue is located in a flexible turn [27,28]. We speculate that no mutations affecting Paxlovid resistance have occurred in this study period. In addition, no mutations were detected in RdRP residues that interact with remdesivir or molnupiravir [29,30].
- Paxlovid has been globally used since it was authorized by the Food and Drug Administration in December 2021. Research has shown that although certain mutations such as P132H, G15S, and K90R have become frequently reported, the binding and inhibitory effects of nirmatrelvir were still not compromised by those variants [31]. Nonetheless, it is obvious that, with the advent of the Omicron variant, the frequency and number of substitutions in the NSP5 region have markedly increased compared to earlier VOCs and VOIs, and the fact that several mutational changes have been occurring at the inhibitor binding sites, albeit at a relatively low frequency [26], indicates that continuous genomic monitoring and characterization of those mutations in the protease sequence should be performed.
- After the Omicron variant emerged in South Korea, we observed a few mutations affecting the detection of the N gene. By carrying these mutations, the virus may have the ability to evade detection, thus having selective advantages such as increased transmission [32]. Recurrence of the mutations in the same genetic region across various lineages during the course of the SARS-CoV-2 pandemic also implies certain beneficial roles of the mutation. The 203−208 del mutation is positioned in the linker region, which links the N-terminal and C-terminal of the N protein. Previous studies have suggested that this deletion might impact phase separation, promoting the function of the region and possibly provides advantages in the packaging and replication process of the virus [33−35]. The N gene was generally considered a highly conserved region and has been used for the target region of several RT-PCR diagnostic kits [36]. However, recent genomic surveillance studies have shown that this gene, particularly the linker region, is one of the least conserved regions in the SARS-CoV-2 genome [33,35,37].
- There also have been reports of similar detection failure problems in several diagnostic kits. A mutation in the E gene (C26340T) was reported to affect E gene detection by the Cobas SARS-CoV-2 assay (Roche Inc., Basel, Switzerland) [38] in 2020. Furthermore, C29200A [39] and G29179T [40] mutations in the N gene were observed to impair the sensitivity of Xpert Xpress SARS-CoV-2 assay (Cepheid Inc., Sunnyvale, CA, USA) in 2020. In early 2021, Zannoli et al. [32] collected VOC B.1.17 specimens showing N gene drop-out using the Allplex SARS-CoV-2 assay, which resulted from the 207−208 deletion or 208-209 indel. Furthermore, more than 100 specimens, mostly Gamma variants, with impaired N gene detection by the GeneFinder Kit were reported by Lesbon et al. [33], and 3 different mutations in the gene were identified as associated mutations, including 202−207 deletion, between April 2020 and July 2021. The constant occurrence of these mutations could affect the diagnostic sensitivity of RT-PCR-based testing and lead to an increase in false-negative results, especially in samples with a low viral load or in single target assays. Therefore, continuous monitoring of the mutations and the consequent sensitivity of various diagnostic kits is crucial for maintaining accurate diagnostics.
- The results provided in this study will contribute to an understanding of the potential impacts of the emerging mutations on evading global efforts to control the virus, thus underlining the need for continual genomic surveillance and characterization of the evolution of SARS-CoV-2. Although whole-genome sequencing plays a key role in pandemic surveillance, the clinical adoption of sequencing has been limited by several aspects, such as higher costs, technical and bioinformatics expertise, and time for implementation. In this respect, the present study showed that utilization of the nanopore sequencing platform and the ARTIC workflow could offer rapid, cost-effective, and reliable sequencing of SARS-CoV-2 samples even at a single hospital. This strategy can serve as a method for increasing the national genomic surveillance capacity for SARS-CoV-2 and other future pathogens in pandemics and epidemics by enabling private labs, academia, and other regional institutions to actively engage in surveillance.
Discussion
Supplementary Material
Table S1.
Table S3.
Table S4.
Figure S1.
-
Ethics Approval
This study was approved by the Institutional Review Board (IRB) of Kangdong Sacred Heart Hospital, Seoul, South Korea (IRB No: NON2021-001-001) and performed in accordance with the principles of the Declaration of Helsinki.
-
Conflicts of Interest
The authors have no conflicts of interest to declare.
-
Funding
This work was supported by Kangdong Sacred Heart Hospital Fund (2021-05) and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI; grant number: HI20C0071), funded by the Ministry of Health & Welfare, Republic of Korea.
-
Availability of Data
The datasets generated during the current study are available in the NCBI GenBank accession no. ON797335-ON797392.
-
Authors’ Contributions
Conceptualization: JSK, HK; Data curation: HK; Formal analysis: HK, HySK; Funding acquisition: JSK; Investigation: SHC, HySK; Methodology: SHC, JSK; Project administration: JSK; Resources: JSK; Software: HK; Supervision: KHH; Validation: HaSK, WS; Visualization: HK; Writing–original draft: HK, JSK, SHC, HySK, HaSK, WS, KHH; Writing–review & editing: all authors.
Article information
- 1. Cucinotta D, Vanelli M. WHO declares COVID-19 a pandemic. Acta Biomed 2020;91:157−60.PubMedPMC
- 2. Kim S, Kim M, Lee S, et al. Discovering spatiotemporal patterns of COVID-19 pandemic in South Korea. Sci Rep 2021;11:24470. ArticlePubMedPMCPDF
- 3. Korea Disease Control and Prevention Agency (KDCA). Domestic COVID-19 mutant virus detection rate in South Korea [Internet]. Cheongju: KDCA; 2022 [cited 2022 Apr 30]. Available from: https://www.kdca.go.kr/contents.es?mid=a20107040000. Korean.
- 4. Amicone M, Borges V, Alves MJ, et al. Mutation rate of SARS-CoV-2 and emergence of mutators during experimental evolution. Evol Med Public Health 2022;10:142−55.ArticlePubMedPMCPDF
- 5. Ma W, Yang J, Fu H, et al. Genomic perspectives on the emerging SARS-CoV-2 Omicron variant. Genomics Proteomics Bioinformatics 2022;20:60−9.Article
- 6. Rochman ND, Wolf YI, Faure G, et al. Ongoing global and regional adaptive evolution of SARS-CoV-2. Proc Natl Acad Sci U S A 2021;118:e2104241118.ArticlePubMedPMC
- 7. O’Toole A, Scher E, Underwood A, et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol 2021;7:veab064. PubMedPMC
- 8. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994;22:4673−80.ArticlePubMedPMC
- 9. Price MN, Dehal PS, Arkin AP. FastTree 2: approximately maximum-likelihood trees for large alignments. PLoS One 2010;5:e9490.ArticlePubMedPMC
- 10. Greaney AJ, Starr TN, Bloom JD. An antibody-escape estimator for mutations to the SARS-CoV-2 receptor-binding domain. Virus Evol 2022;8:veac021. ArticlePubMedPMCPDF
- 11. Aksamentov I, Roemer C, Hodcroft EB, et al. Nextclade: clade assignment, mutation calling and quality control for viral genomes. J Open Source Softw 2021;6:3773. ArticlePDF
- 12. Wang Q, Zhang Y, Wu L, et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020;181:894−904.ArticlePubMedPMC
- 13. Zhang L, Jackson CB, Mou H, et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat Commun 2020;11:6013. ArticlePubMedPMCPDF
- 14. Teng S, Sobitan A, Rhoades R, et al. Systemic effects of missense mutations on SARS-CoV-2 spike glycoprotein stability and receptor-binding affinity. Brief Bioinform 2021;22:1239−53.ArticlePubMed
- 15. Genovese L, Zaccaria M, Farzan M, et al. Investigating the mutational landscape of the SARS-CoV-2 Omicron variant via ab initio quantum mechanical modeling [Preprint]. Posted 2021 Dec 3. BioRxiv 2021.12.01.470748. https://doi.org/10.1101/2021.12.01.470748.Article
- 16. Yin W, Xu Y, Xu P, et al. Structures of the Omicron spike trimer with ACE2 and an anti-Omicron antibody. Science 2022;375:1048−53.ArticlePubMedPMC
- 17. Carter C, Airas J, Parish CA. Wild type and Omicron SARS-CoV-2 spike receptor binding domains bind similarly to the human ACE2 receptor: an MM-GBSA Study [Preprint]. Posted 2022 Jan 7. ChemRxiv 2022-30zcn. https://doi.org/10.26434/chemrxiv-2022-30zcn.Article
- 18. Motozono C, Toyoda M, Tan TS, et al. The SARS-CoV-2 Omicron BA. 1 spike G446S potentiates HLA-A*24:02-restricted T cell immunity [Preprint]. Posted 2022 Apr 18. bioRxiv 2022.04.17.488095. https://doi.org/10.1101/2022.04.17.488095.Article
- 19. Sarkar R, Lo M, Saha R, et al. S glycoprotein diversity of the Omicron variant [Preprint]. Posted 2021 Dec 6. MedRxiv 2021.12.04.21267284. https://doi.org/10.1101/2021.12.04.21267284.Article
- 20. Liu Z, VanBlargan LA, Bloyet LM, et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021;29:477−88.ArticlePubMedPMC
- 21. Greaney AJ, Loes AN, Crawford KH, et al. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021;29:463−76.ArticlePubMedPMC
- 22. Tada T, Zhou H, Dcosta BM, et al. Increased resistance of SARS-CoV-2 Omicron variant to neutralization by vaccine-elicited and therapeutic antibodies. EBioMedicine 2022;78:103944. ArticlePubMedPMC
- 23. Sharma V, Rai H, Gautam DN, et al. Emerging evidence on Omicron (B.1.1.529) SARS-CoV-2 variant. J Med Virol 2022;94:1876−85.ArticlePubMedPMCPDF
- 24. Andreano E, Piccini G, Licastro D, et al. SARS-CoV-2 escape from a highly neutralizing COVID-19 convalescent plasma. Proc Natl Acad Sci U S A 2021;118:e2103154118.ArticlePubMedPMC
- 25. Lee JT, Yang Q, Gribenko A, et al. Genetic surveillance of SARS-CoV-2 Mpro reveals high sequence and structural conservation prior to the introduction of protease inhibitor Paxlovid. mBio 2022;13:e0086922.ArticlePubMedPMCPDF
- 26. Motyan JA, Mahdi M, Hoffka G, et al. Potential resistance of SARS-CoV-2 main protease (Mpro) against protease inhibitors: lessons learned from HIV-1 protease. Int J Mol Sci 2022;23:3507. ArticlePubMedPMC
- 27. Sacco MD, Hu Y, Gongora MV, et al. The P132H mutation in the main protease of Omicron SARS-CoV-2 decreases thermal stability without compromising catalysis or small-molecule drug inhibition. Cell Res 2022;32:498−500.ArticlePubMedPMCPDF
- 28. Vangeel L, Chiu W, De Jonghe S, et al. Remdesivir, Molnupiravir and Nirmatrelvir remain active against SARS-CoV-2 Omicron and other variants of concern. Antiviral Res 2022;198:105252. ArticlePubMedPMC
- 29. Stevens LJ, Pruijssers AJ, Lee HW, et al. Mutations in the SARS-CoV-2 RNA-dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci Transl Med 2022;14:eabo0718.ArticlePubMedPMC
- 30. Padhi AK, Shukla R, Saudagar P, et al. High-throughput rational design of the remdesivir binding site in the RdRp of SARS-CoV-2: implications for potential resistance. iScience 2020;24:101992. ArticlePubMedPMC
- 31. Ullrich S, Ekanayake KB, Otting G, et al. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg Med Chem Lett 2022;62:128629. ArticlePubMedPMC
- 32. Zannoli S, Dirani G, Taddei F, et al. A deletion in the N gene may cause diagnostic escape in SARS-CoV-2 samples. Diagn Microbiol Infect Dis 2022;102:115540. ArticlePubMedPMC
- 33. Lesbon JC, Poleti MD, de Mattos Oliveira EC, et al. Nucleocapsid (N) gene mutations of SARS-CoV-2 can affect real-time RT-PCR diagnostic and impact false-negative results. Viruses 2021;13:2474. ArticlePubMedPMC
- 34. Mauger DM, Cabral BJ, Presnyak V, et al. mRNA structure regulates protein expression through changes in functional half-life. Proc Natl Acad Sci U S A 2019;116:24075−83.ArticlePubMedPMC
- 35. Lu S, Ye Q, Singh D, et al. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat Commun 2021;12:502. ArticlePubMedPMC
- 36. Dutta NK, Mazumdar K, Gordy JT. The nucleocapsid protein of SARS-CoV-2: a target for vaccine development. J Virol 2020;94:e00647−20.ArticlePubMedPMCPDF
- 37. Wang R, Hozumi Y, Yin C, et al. Mutations on COVID-19 diagnostic targets. Genomics 2020;112:5204−13.ArticlePubMedPMC
- 38. Artesi M, Bontems S, Gobbels P, et al. A recurrent mutation at position 26340 of SARS-CoV-2 is associated with failure of the E gene quantitative reverse transcription-PCR utilized in a commercial dual-target diagnostic assay. J Clin Microbiol 2020;58:e01598−20.ArticlePubMedPMCPDF
- 39. Hasan MR, Sundararaju S, Manickam C, et al. A novel point mutation in the N gene of SARS-CoV-2 may affect the detection of the virus by reverse transcription-quantitative PCR. J Clin Microbiol 2021;59:e03278−20.ArticlePubMedPMCPDF
- 40. Hong KH, In JW, Lee J, et al. Prevalence of a single-nucleotide variant of SARS-CoV-2 in Korea and its impact on the diagnostic sensitivity of the Xpert Xpress SARS-CoV-2 assay. Ann Lab Med 2022;42:96−9.ArticlePubMedPMC
References
Figure & Data
References
Citations
- Understanding large scale sequencing datasets through changes to protein folding
David Shorthouse, Harris Lister, Gemma S Freeman, Benjamin A Hall
Briefings in Functional Genomics.2024;[Epub] CrossRef - Molecular epidemiology of SARS‐CoV‐2 in Mongolia, first experience with nanopore sequencing in lower‐ and middle‐income countries setting
Munkhtuya Erendereg, Suvd Tumurbaatar, Otgonjargal Byambaa, Gerelmaa Enebish, Natsagdorj Burged, Tungalag Khurelsukh, Nomin‐Erdene Baatar, Badmaarag Munkhjin, Jargaltulga Ulziijargal, Anuujin Gantumur, Oyunbaatar Altanbayar, Ochbadrakh Batjargal, Delgermu
Immunity, Inflammation and Disease.2023;[Epub] CrossRef
 PubReader
PubReader Cite
Cite