Genomic Surveillance of SARS-CoV-2: Distribution of Clades in the Republic of Korea in 2020
Article information
Abstract
Since a novel beta-coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first reported in December 2019, there has been a rapid global spread of the virus. Genomic surveillance was conducted on samples isolated from infected individuals to monitor the spread of genetic variants of SARS-CoV-2 in Korea. The Korea Disease Control and Prevention Agency performed whole genome sequencing of SARS-CoV-2 in Korea for 1 year (January 2020 to January 2021). A total of 2,488 SARS-CoV-2 cases were sequenced (including 648 cases from abroad). Initially, the prevalent clades of SARS-CoV-2 were the S and V clades, however, by March 2020, GH clade was the most dominant. Only international travelers were identified as having G or GR clades, and since the first variant 501Y.V1 was identified (from a traveler from the United Kingdom on December 22nd, 2020), a total of 27 variants of 501Y.V1, 501Y.V2, and 484K.V2 have been classified (as of January 25th, 2021). The results in this study indicated that quarantining of travelers entering Korea successfully prevented dissemination of the SARS-CoV-2 variants in Korea.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first reported in December 2019, in Wuhan, China, and subsequently, the virus has spread rapidly all over the world [1]. Since the first novel coronavirus genome sequence was made available (January 10th, 2020 [2]), whole genome sequencing (WGS) has been extensively performed globally, to characterize the virus. This has led to many important findings, and more than 412,699 complete genome sequences have been made available in a global initiative on sharing all influenza data (GISAID [as of January 25th, 2021]) [3]. Detection of novel variants of SARS-CoV-2 including 501Y.V1 (also known as Variant of Concern 2020/12/01) [4], 501Y.V2 [5] and 484K.V2 [6], which have rapidly spread worldwide, have emphasized the need for sequence-based strain surveillance, to promptly detect mutations to prevent the spread of new variants.
The Republic of Korea was one of the first countries to report a significant Coronavirus Disease-19 (COVID-19) outbreak which was managed successfully due to the effective cooperation between the government and the people of Korea. As part of a comprehensive national surveillance of COVID-19, the Korea Disease Control and Prevention Agency (KDCA) conducted the WGS of SARS-CoV-2 using real-time genomic epidemiology data to monitor how the virus mutates, and correlates with sequence variation since the first case of SARS-CoV-2 infection was identified in Korea on January 20th, 2020 [7].
In this Brief Report, the SARS-CoV-2 genomes of 2,488 confirmed cases in Korea were sequenced and analyzed, in the context of the global viral population, from the beginning of the pandemic to provide valuable insights into the genetic pattern of SARS-CoV-2 distribution in Korea.
Materials and Methods
Nasopharyngeal and oropharyngeal swabs were collected from SARS-CoV-2, real-time reverse transcription (RT)-PCR confirmed cases. RNA extraction and RT-PCR were performed on the samples from the swabs, according to the methods described in a previous report [8].
To perform WGS, cDNA was amplified using primer pools (https://artic.network/ncov-2019). Libraries were prepared using the Nextera DNA Flex Library Prep Kit (Illumina, USA), and sequenced on the MiSeq instrument (Illumina, USA) with 2 × 150 base pairs using a MiSeq reagent kit V2 (Illumina, USA) to obtain an average genome coverage greater than 1,000 × for all the isolates. The sequence reads were trimmed and mapped to reference genome MN908947.3 using CLC Genomics Workbench Version 20.0.3 (CLC Bio, Denmark).
All types of clades (including variants) were isolated by the KDCA (Division of Emerging Infectious Diseases) using the Vero E6 cell line and were deposited in the National Culture Collection for Pathogens and sequence data were uploaded to GISAID (Table 1).

Lists of 7 clades and 3 variants of SARS-CoV-2.
The study was approved by the International Review Board at the Korea Disease Control and Prevention Agency (2020-03-01-P-A). The board waived the requirement for written consent.
Results
1. SARS-CoV-2 genomic surveillance in Korea
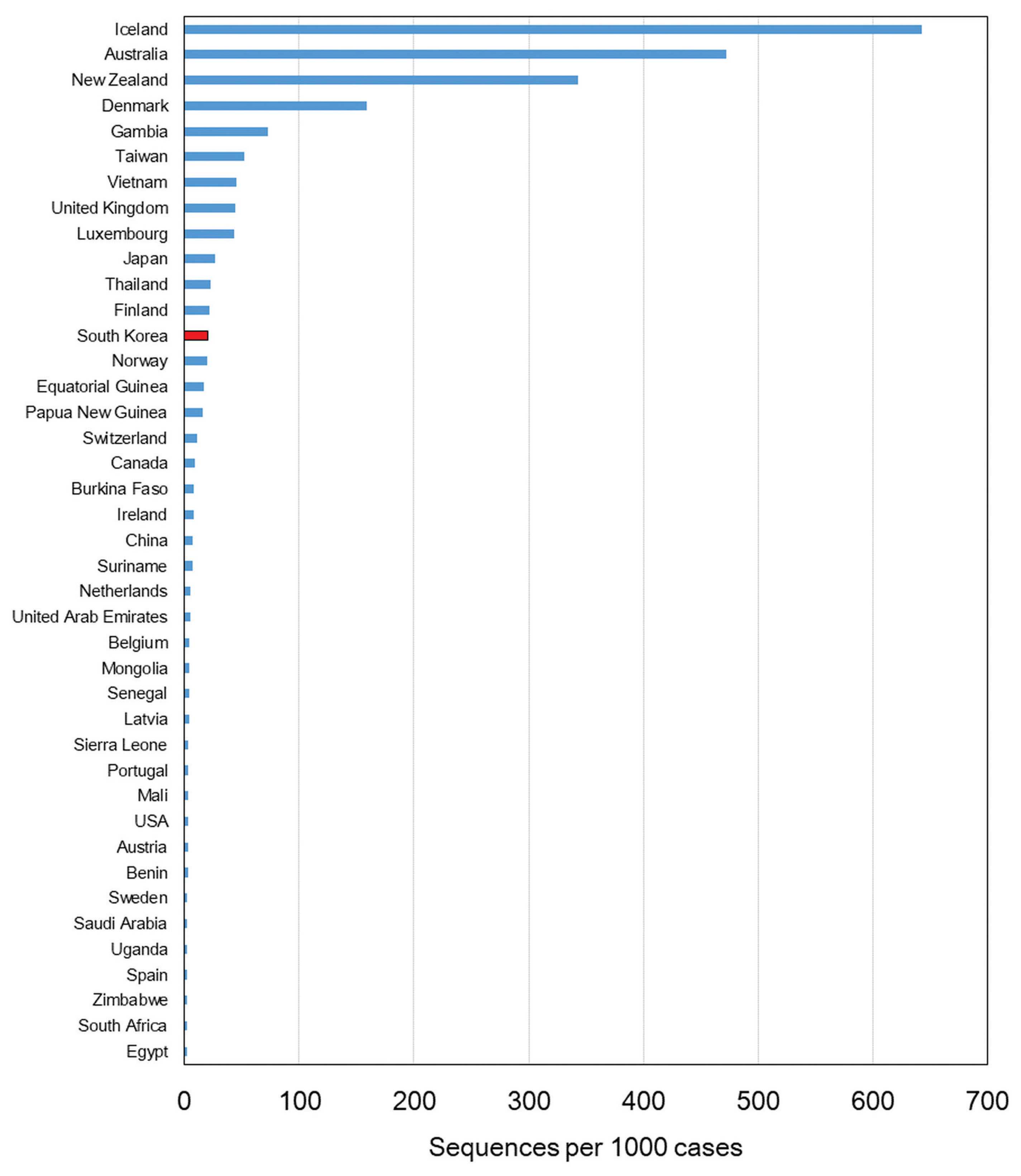
Since the first confirmed case reported on January 20th, 2020, the number of confirmed cases has reached 75,521 as of January 25th, 2021. The number of cases imported from abroad were 6,144. The daily number of reported COVID-19 cases in Korea is shown in Figure 1. The first wave of COVID-19, which was associated with the Shincheonji outbreak, began in February and lasted until April 2020 [9]. In May 2020, another outbreak of COVID-19 was reported which was traced to the Itaewon district in Seoul. A second wave of COVID-19 in Korea began in August 2020, and a third wave in November 2020, both originating in the Seoul area. As indicated by the number of sequenced genomes, WGS in Korea has covered more than 27% of the total number of confirmed cases as of May 2020. There was a remarkable increase in confirmed cases during the third wave which resulted in a decrease of sequencing during this period, but overall, the sequencing of SARS-CoV-2 in Korea was about 3.3% of all COVID-19 confirmed cases. Based on the percentage of COVID-19 cases which were genome sequenced, and uploaded to GISAID, Korea ranked in the top 15 countries (Figure 2).

Time series of new daily confirmed cases of COVID-19 in Korea from January 2020 to January 25, 2021 (blue). The orange line represents the number sequenced genomes per month. Sequencing of SARS-CoV-2 for January 2021 is in progress.
2. Distribution of clades in Korea
A total of 2,488 SARS-CoV-2 were sequenced during the period between January 20th, 2020 and January 25th (2,021 including 648 cases imported from abroad). The overall distribution of clades over time is shown in Figure 3A. There was an initial period during January to March 2020 when the S and V clades (GISAID nomenclatures) were more prevalent in total than G/GR/GH clades in Korea (Figure 3B). S and V clades were the most prevalent clades at the beginning of the epidemic however, after the emergence of the variant D614G spike protein mutation, this variant became predominant [10]. The G/GR/GH clades were first identified in March 2020 in Korea, with the GH clade becoming dominant. Interestingly, the G/GR clades identified in Korea, were only isolated from international travelers, and the GR clades were only isolated from cases in close contact with someone who came from abroad indicating there was a low prevalence of GR in Korea. The diversity of clades observed in cases imported from abroad compared with domestic cases, led to the belief that COVID-19 from abroad had been restricted through comprehensive, and strict quarantine and disease control measures (Figure 3B). A comparison of the prevalent clades in each continent highlights a higher prevalence of the GH clade in Korea (Figure 4).
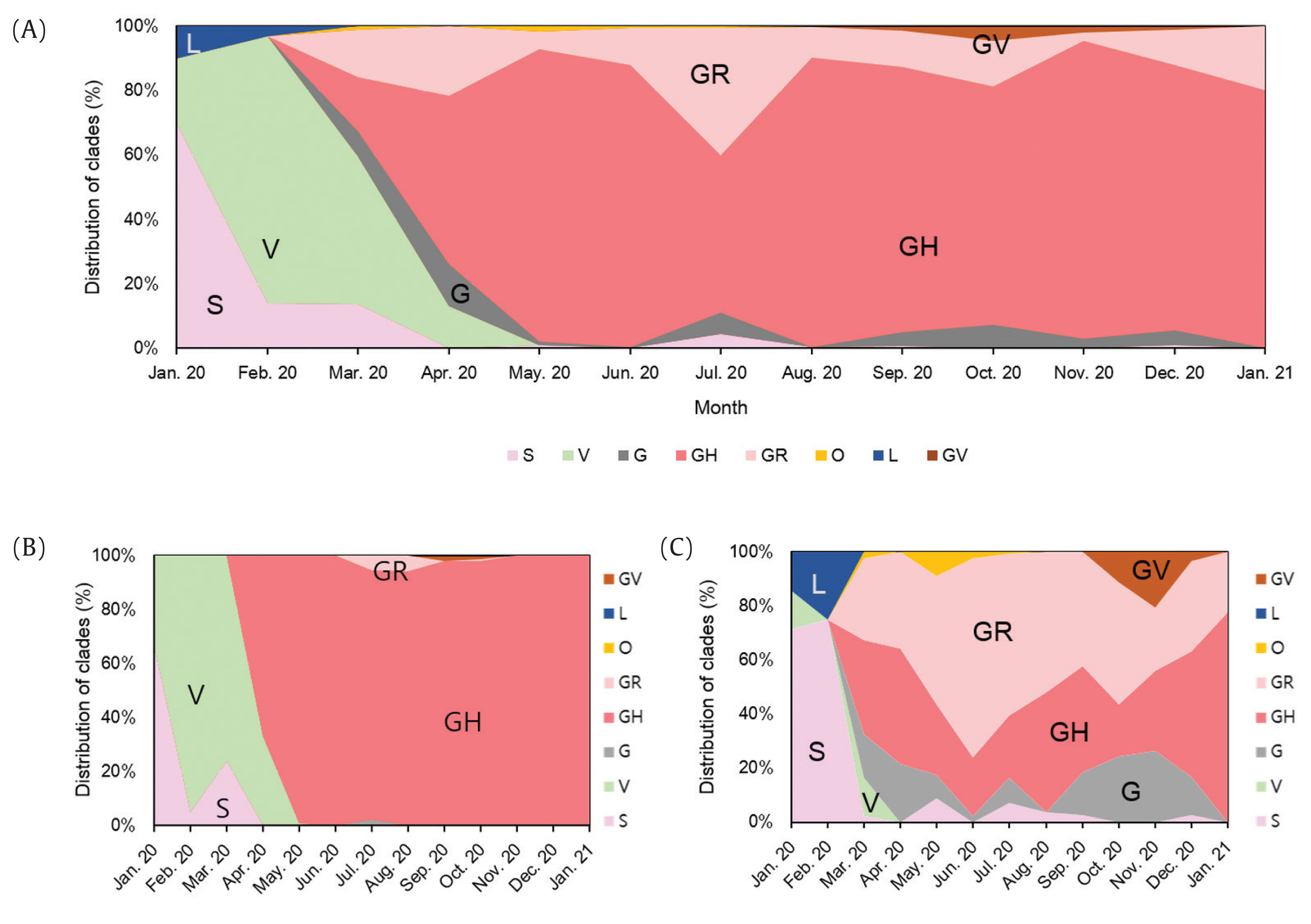
Distribution of total, domestic, and imported SARS-CoV-2 clades in Korea. (A) Total clades - A total of 2,488 SARS-CoV-2 samples were sequenced. (B) Domestic clades - A total of 1,840 cases of SARS-CoV-2 were attributed to domestic cases. Note that majority of clades were classified into GH. (C) Foreign clades - A total of 648 cases of SARS-CoV-2 were imported from abroad. The clade composition of cases imported from abroad was more diverse than domestic cases.
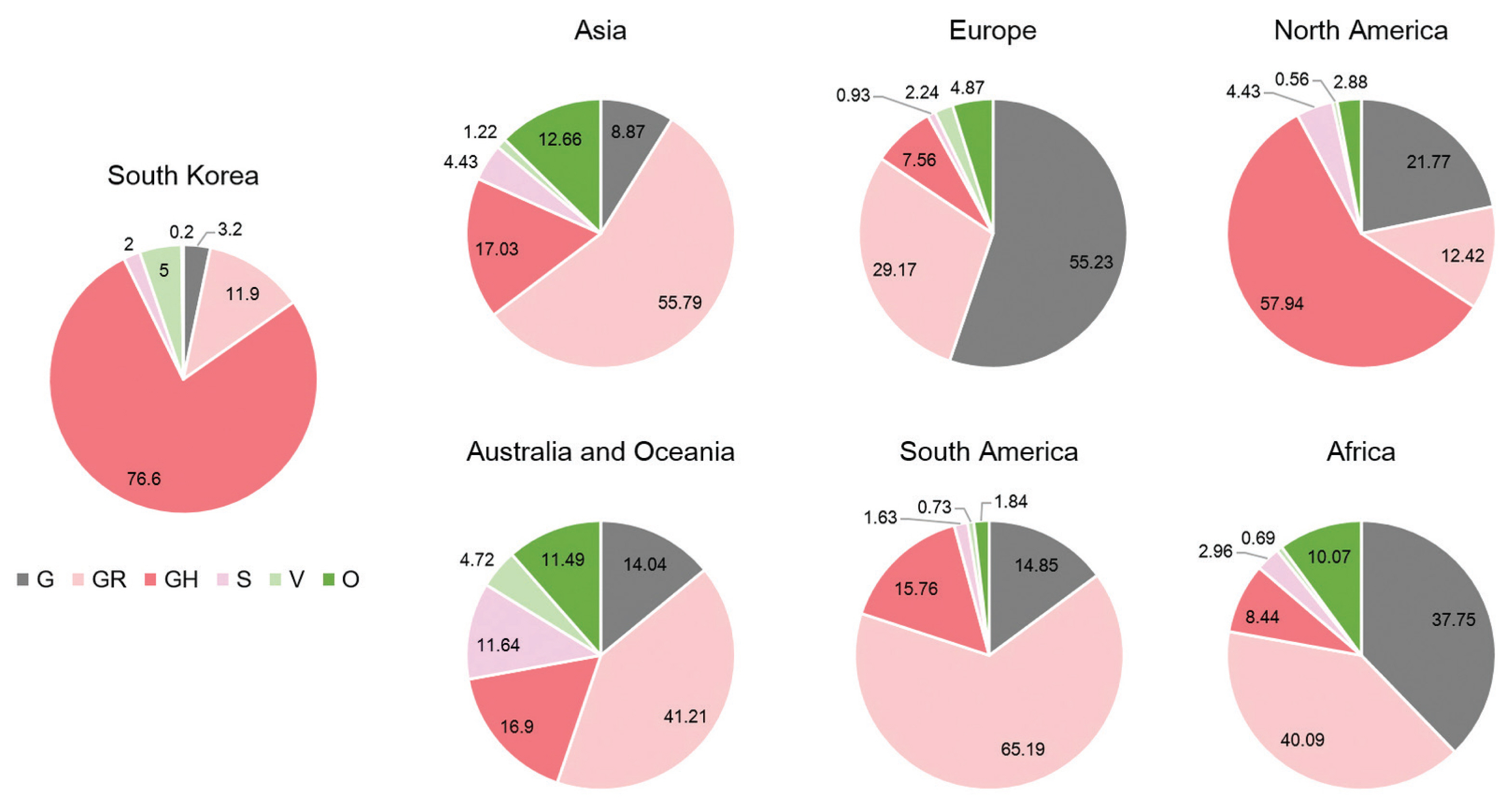
Distribution of clades by continents. The proportion of clades observed in each continent was calculated using all available sequences from the GISAID.
GISAID = global initiative on sharing all influenza data.
The analysis of domestic cases of SARS-CoV-2 in Korea using the Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN v2.1.7) database [11] indicated that over 80% of isolates were classified as B.1.3.1 (as of January 25th, 2021). The proportion of the B.1.3.1 lineage has continued to rise steadily as shown in Figure 5. Analysis of domestic cases also supports that SARS-CoV-2 imported from abroad does not seem to be spreading within Korea because the lineage distributions was the same (B.1.3.1) from May 2020 to January 2021.

Analysis of the PANGOLIN database lineage isolated from domestic cases of SARS-CoV-2 in Korea. The majority of isolates were classified as B.1.3.1.
PANGOLIN = Phylogenetic Assignment of Named Global Outbreak Lineages.
3. Multiple SARS-CoV-2 variants
Multiple SARS-CoV-2 variants have been circulating globally. The main variants include 501Y.V1 (B.1.1.7), 501Y.V2 (B.1.351), and 484K.V2 (P.1 and P.2). The nomenclatures in parentheses are assigned by the PANGOLIN database which was developed by members of the COVID-19 Genomics UK team [11]. The PANGOLIN database was deployed to establish the transmission patterns of numerous variants of the virus. The KDCA has carried out investigations into travelers who returned from abroad. The first 501Y.V1 variant was identified as originating from a person traveling from the United Kingdom on December 22nd, 2020, and an additional 18 cases were identified as of January 25th, 2021 (Table 1).
The first 501Y.V2 variant was identified from a person traveling from South Africa on December 26th, 2020, and 4 additional cases were identified as of January 25th, 2021 (Table 1). The first 484K.V2 was identified from a person traveling from Brazil on January 10th, 2021, and 2 additional cases were confirmed as of January 25th, 2021 (Table 1).
Among 19 cases of 501Y.V1, 2 were identified from people traveling from the Maldives and Ghana, on January 25th, 2021. Among 5 cases of 501Y.V2, 3 were confirmed from people traveling from Zimbabwe, Malawi, and Tanzania on January 25th, 2021. Through surveillance, 501Y.V1 was first identified from a person traveling from the Maldives, and 501Y.V2 from two people traveling from Zimbabwe and Malawi.
Discussion
In January 2020, following the first case of COVID-19 in Korea, real-time genomic surveillance was being conducted by the KDCA. Based on the percentage of COVID-19 cases which were genome sequenced, and uploaded to GISAID (1,632 as of January 25th, 2021), Korea ranked in the top 15 countries.
Analysis of genome sequences indicated that from January to March 2020, S and V were the prevalent clades in Korea. However, the prevalent clade of Korea is now GH (first isolated in March 2020). The other clades which are prevalent in Europe, G and GR, have not been identified in domestic cases of COVID-19. In addition, analysis of the PANGOLIN database lineages indicated that the majority of SARS-CoV-2 in Korea were grouped into the B.1.3.1 lineage. Therefore, these results indicated that quarantine for all travelers entering Korea played a crucial role in preventing the variants of SARS-CoV-2 spreading in Korea.
Furthermore, genomic surveillance has also played a crucial role in monitoring recent variants including 501Y.V1, 501Y.V2, and 484K.V2. To date these variants of SARS-CoV-2 have not been introduced and spread in Korea, indicating that the quarantine and containment strategy was effective.
Despite several sharp spikes in the number of confirmed COVID-19 cases in Korea in 2020, the cooperation between the Korean government and the general population has enabled a successful 3T strategy (Test, Trace, and Treat) for containing the COVID-19 pandemic. The KDCA will continue to contribute to a global effort to better understand, treat, and prevent COVID-19.
Acknowledgements
We are grateful to all the contributions from the laboratories responsible for obtaining the specimens and all the laboratories where genetic sequence data were generated, and shared via the GISAID, on which this research is based. Special thanks to the Emergency Operation Center of the KDCA for its great effort in recording new confirmed cases.
This study was funded by Korea Disease Control and Prevention Agency (no.: 4800-4837-301, 4800-4834-303).
Notes
Conflicts of Interest
The authors have no conflicts of interest to declare.